
Sophia Vincoff
commited on
Commit
·
bae913a
1
Parent(s):
8d9d9da
caid benchmark
Browse filesThis view is limited to 50 files because it contains too many changes.
See raw diff
- fuson_plm/benchmarking/caid/README.md +294 -0
- fuson_plm/benchmarking/caid/__init__.py +0 -0
- fuson_plm/benchmarking/caid/analyze_fusion_preds.py +158 -0
- fuson_plm/benchmarking/caid/clean.py +671 -0
- fuson_plm/benchmarking/caid/color_disordered_residues.ipynb +849 -0
- fuson_plm/benchmarking/caid/config.py +14 -0
- fuson_plm/benchmarking/caid/disorder_coloring_data/normalized_disorder_propensities_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/model.py +26 -0
- fuson_plm/benchmarking/caid/plot.py +1030 -0
- fuson_plm/benchmarking/caid/process_fusion_structures.py +799 -0
- fuson_plm/benchmarking/caid/processed_data/CAID-2_Disorder_NOX_Processed.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/IDP-CRF_Training_Dataset.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/AlphaFold-disorder_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/AlphaFold-rsa_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/DISOPRED3-diso_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/DeepIDP-2L_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/DisoPred_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/Dispredict3_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/ESpritz-D_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/IDP-Fusion_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/IUPred3_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/disomine_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPlr2_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPnn2_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPnn_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPtr_CAID-2_Disorder_NOX.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EML4-ALK.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EML4::ALK_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EWSR1-FLI1.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EWSR1::FLI1_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_PAX3-FOXO1.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_PAX3::FOXO1_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_SS18-SSX1.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_SS18::SSX1_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/disorder_nox_histogram.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/disorder_nox_histogram_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/fusions_histogram.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/fusions_histogram_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/heads_histogram.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/heads_histogram_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/tails_histogram.png +0 -0
- fuson_plm/benchmarking/caid/processed_data/figures/histograms/tails_histogram_source_data.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/flDPnn_Training_Dataset.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/flDPnn_Validation_Dataset.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/fusionpdb/FusionPDB_level2-3_cleaned_FusionGID_info.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/fusionpdb/fusion_heads_and_tails.csv +3 -0
- fuson_plm/benchmarking/caid/processed_data/fusionpdb/heads_tails_structural_data.csv +3 -0
- fuson_plm/benchmarking/caid/raw_data/caid2_competition_results/AlphaFold-disorder.caid +0 -0
- fuson_plm/benchmarking/caid/raw_data/caid2_competition_results/AlphaFold-rsa.caid +0 -0
fuson_plm/benchmarking/caid/README.md
ADDED
|
@@ -0,0 +1,294 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
## CAID Benchmark
|
| 2 |
+
|
| 3 |
+
This folder contains all the data and code needed to perform the **CAID benchmark**, where FusOn-pLM-Diso (a classifier built on FusOn-pLM embeddings) is used to predict per-residue disorder propensities (Figure 4C-F) and plot disorder properties (Figure 1C-1D, S1)
|
| 4 |
+
|
| 5 |
+
### TL;DR
|
| 6 |
+
The order in which to run the scripts:
|
| 7 |
+
|
| 8 |
+
```
|
| 9 |
+
python scrape_fusionpdb.py # pull FusionPDB structures
|
| 10 |
+
python process_fusion_structures.py # process FusionPDB structures, and head/tail protein structures
|
| 11 |
+
python clean.py # clean disorder data and structure data. Assemble train/test/benchmark splits
|
| 12 |
+
python train.py # train models
|
| 13 |
+
python analyze_fusion_preds.py # make box chart and line plot of model performance on fusion proteins
|
| 14 |
+
python plot.py # plot AUROC of model performance, and additional figures based on disorder data
|
| 15 |
+
```
|
| 16 |
+
|
| 17 |
+
Additional notes:
|
| 18 |
+
|
| 19 |
+
* `color_disorder_residues.ipynb` is used to plot fusion structures with pLDDT or disorder prediction color overlays.
|
| 20 |
+
* We recommend using `nohup` to run longer scripts like `scrape_fusionpdb.py`, `process_fusion_structures.py`, `clean.py`, and `train.py`
|
| 21 |
+
|
| 22 |
+
### Downloading raw disorder data
|
| 23 |
+
Per-residue disorder predictions were used to train and test FusOn-pLM-Diso.
|
| 24 |
+
|
| 25 |
+
1. **flDPnn** ([Hu et al. 2021](https://doi.org/10.1038/s41467-021-24773-7))
|
| 26 |
+
1. At this [link](http://biomine.cs.vcu.edu/servers/flDPnn/?fbclid=IwZXh0bgNhZW0CMTEAAR0KO5CkNdkGC9e5O32S0QoG3BWOw6_egbnioXQNBSv3UC-m_b_dxh70Nnk_aem_z285WFCHdBLw3vOj7LL37A), scroll down to the bottom to find links to the [training](http://biomine.cs.vcu.edu/servers/flDPnn/data/flDPnn_Training_Annotation.txt) and [validation](http://biomine.cs.vcu.edu/servers/flDPnn/data/flDPnn_Validation_Annotation.txt) sets.
|
| 27 |
+
2. **IDP-CRF** ([Liu et al. 2018](https://doi.org/10.3390/ijms19092483))
|
| 28 |
+
1. Download zipped data from [this link](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6164615/bin/ijms-19-02483-s001.zip), remove header and footer, and save as a FASTA file
|
| 29 |
+
3. **CAID2-Disorder-NOX** ([Del Conte et al. 2023](https://doi.org/10.1002/prot.26582))
|
| 30 |
+
1. Go to [CAID Round 2 Results](https://caid.idpcentral.org/challenge/results?fbclid=IwZXh0bgNhZW0CMTEAAR12dKaA0KywcT71FnyXIrrNS91pwGREsLiq5c2RmfdYl7L0VdUNG7jYai8_aem_tW6Wm9_11ZuiI_GKzbNZjA). Scroll to "Here you can download the references used in the CAID-2 challenge" and you'll find the following links.
|
| 31 |
+
1. [disorder_nox.fasta](https://caid.idpcentral.org/assets/sections/challenge/static/references/2/disorder_nox.fasta)
|
| 32 |
+
2. [predictions](https://caid.idpcentral.org/assets/sections/challenge/static/predictions/2/predictions.zip) made by all CAID2 participants; AUROC curves can be reconstructed from these
|
| 33 |
+
|
| 34 |
+
Raw disorder data are stored in `caid/raw_data`
|
| 35 |
+
```
|
| 36 |
+
benchmarking/
|
| 37 |
+
└── caid/
|
| 38 |
+
└── raw_data/
|
| 39 |
+
└── caid2_competition_results/...
|
| 40 |
+
└── caid2_train_and_test_data/
|
| 41 |
+
├── CAID-2_Disorder_NOX_Testing_Sequences.fasta
|
| 42 |
+
├── flDPnn_Training_Dataset.txt
|
| 43 |
+
├── flDPnn_Validation_Annotation.txt
|
| 44 |
+
├── IDP-CRF_Training_Dataset.txt
|
| 45 |
+
```
|
| 46 |
+
- 📁 **`raw_data/caid2_competition_results/`**: folder containing raw predictions from CAID2 competitors, downloaded directly from the CAID2 website. Models: AlphaFold-disorder, AlphaFOld-rsa, DeepIDP-2L, disomine, DisoPred, DISOPRED3-diso, Dispredict3, ESpritz-D, flDPlr2, flDPnn, flDPnn2, flDPtr, IDP-Fusion, IUPred3.
|
| 47 |
+
- **`raw_data/caid2_train_and_test_data/CAID-2_Disorder_NOX_Testing_Sequences.fasta`**: Disorder-NOX dataset (used as the test set in this benchmark)
|
| 48 |
+
- **`raw_data/caid2_train_and_test_data/flDPnn_Training_Dataset.txt`**: training set for flDPnn
|
| 49 |
+
- **`raw_data/caid2_train_and_test_dataflDPnn_Validation_Dataset.txt`**: validation set for flDPnn
|
| 50 |
+
- **`raw_data/IDP-CRF_Training_Dataset.txt`**: training set for IDP-CRF
|
| 51 |
+
|
| 52 |
+
### Processing disorder data
|
| 53 |
+
|
| 54 |
+
```
|
| 55 |
+
benchmarking/
|
| 56 |
+
└── caid/
|
| 57 |
+
└── processed_data/
|
| 58 |
+
└── caid2_competition_results/...
|
| 59 |
+
├── CAID-2_Disorder_NOX_Processed.csv
|
| 60 |
+
├── flDPnn_Training_Dataset.csv
|
| 61 |
+
├── flDPnn_Validation_Dataset.csv
|
| 62 |
+
├── IDP-CRF_Training_Dataset.csv
|
| 63 |
+
└── splits/
|
| 64 |
+
├── splits.csv
|
| 65 |
+
├── train_df.csv
|
| 66 |
+
├── test_df.csv
|
| 67 |
+
├── fusion_bench_df.csv
|
| 68 |
+
```
|
| 69 |
+
|
| 70 |
+
The **`clean.py`** processes and combines the raw data files, generating the following files in 📁`processed_data/`:
|
| 71 |
+
- 📁 **`caid2_competition_results/`**: a folder with table versions of all the files in 📁 `raw_data/caid2_competition_results/`
|
| 72 |
+
- **`CAID-2_Disorder_NOX_Processed.csv`**: a table of test data, made by parsing `raw_data/caid2_train_and_test_data/CAID-2_Disorder_NOX_Testing_Sequences.fasta`
|
| 73 |
+
- **`flDPnn_Training_Dataset.csv`**: a table of flDPnn's training data, made by parsing `raw_data/caid2_train_and_test_data/flDPnn_Training_Dataset.txt`
|
| 74 |
+
- **`flDPnn_Validation_Dataset.csv`**: a table of flDPnn's validation data, made by parsing `raw_data/caid2_train_and_test_data/flDPnn_Validation_Dataset.txt`
|
| 75 |
+
- **`IDP-CRF_Training_Dataset.csv`**: a table of IDP-CRF's training data, made by parsing `raw_data/caid2_train_and_test_data/CRF_Training_Dataset.txt`
|
| 76 |
+
|
| 77 |
+
`clean.py` also generates **the final train-test splits and fusion oncoprotein benchmarking file used to train and evaluate the disorder predictors.** These are stored in 📁`splits/`
|
| 78 |
+
- **`splits.csv`**: sequences, IDs, split (either "Train", "Test", or "Fusion_Benchmark"), andpper-residue disorder labels based on AlphaFold-pLDDT (1 (disordered) if pLDDT< 68.8, 0 (ordered) if >=68.8)
|
| 79 |
+
- **`train_df.csv`**: just the Train set portion of `splits.csv`
|
| 80 |
+
- **`test_df.csv`**: just the Test set portion of `splits.csv`
|
| 81 |
+
- **`fusion_bench_df.csv`**: just the Fusion_Benchmark portion of `splits.csv`. Includes 524 fusion oncoproteins from the FusOn-pLM test set whose structures were collected from FusionPDB (see "Downloading and Processing FusionPDB data
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
### Downloading and Processing FusionPDB data
|
| 85 |
+
The structures of fusion oncoproteins from the FusionPDB database were used to evaluate FusOn-pLM-Diso's performance on fusion oncoproteins. This data was collected by running `scrape_fusionpdb.py`, followed by `process_fusion_structures.py`. These scripts populated the `raw_data` and `processed_data` files simultaneously.
|
| 86 |
+
|
| 87 |
+
Listed below are all the relevant files:
|
| 88 |
+
|
| 89 |
+
```
|
| 90 |
+
benchmarking/
|
| 91 |
+
└── caid/
|
| 92 |
+
└── raw_data/
|
| 93 |
+
└── fusionpdb/
|
| 94 |
+
└── structures/... # created by scrape_fusionpdb.py (folder not included in repo)
|
| 95 |
+
└── head_tail_af2db_structures/... # created by process_fusion_structures.py (folder not included in repo)
|
| 96 |
+
├── FusionPDB_level2_curated_09_05_2024.csv
|
| 97 |
+
├── FusionPDB_level2_fusion_structure_links.csv
|
| 98 |
+
├── FusionPDB_level3_curated_09_05_2024.csv
|
| 99 |
+
├── FusionPDB_level3_fusion_structure_links.csv
|
| 100 |
+
├── fusionpdb_structureless_ids.txt
|
| 101 |
+
├── hgene_tgene_uniprot_idmap_07_10_2024.txt
|
| 102 |
+
├── level2_head_tail_info.txt
|
| 103 |
+
├── level3_head_tail_info.txt
|
| 104 |
+
├── not_in_afdb_idmap.txt
|
| 105 |
+
└── processed_data/
|
| 106 |
+
└── fusion_pdb/
|
| 107 |
+
└── intermediates/
|
| 108 |
+
├── giant_level_2-3_fusion_protein_head_tail_info.csv
|
| 109 |
+
├── giant_level2-3_fusion_protein_structure_links.csv
|
| 110 |
+
├── giant_level2-3_fusion_protein_structures_processed.csv
|
| 111 |
+
├── uniprotids_not_in_afdb.txt
|
| 112 |
+
├── unmapped_parts.tt
|
| 113 |
+
├── fusion_heads_and_tails.csv
|
| 114 |
+
├── FusionPDB_level2-3_cleaned_FusionGID_info.csv
|
| 115 |
+
├── FusionPDB_level2-3_cleaned_structure_info.csv
|
| 116 |
+
├── heads_tails_structural_data.csv
|
| 117 |
+
```
|
| 118 |
+
|
| 119 |
+
#### ⚙️ Pipeline
|
| 120 |
+
|
| 121 |
+
Here we describe what each script does and which files each script creates.
|
| 122 |
+
1. 🐍 **`scrape_fusionpdb.py`**
|
| 123 |
+
1. Scrapes metadata for FusionPDB Level 2 and Level 3
|
| 124 |
+
1. Pulls the online tables for [Level 2](https://compbio.uth.edu/FusionPDB/gene_search_result_0.cgi?type=chooseLevel&chooseLevel=level2) and [Level 3](https://compbio.uth.edu/FusionPDB/gene_search_result_0.cgi?type=chooseLevel&chooseLevel=level3), saving results to `raw_data/FusionPDB_level2_curated_09_05_2024.csv` and `raw_data/FusionPDB_level3_curated_09_05_2024.csv` respectively.
|
| 125 |
+
2. Retrieves structure links
|
| 126 |
+
1. Using the tables collected in step (i), visits the page for each fusion oncoprotein (FO) in FusionPDB Level 2 and 3, and downloads all AlphaFold2 structure links for each FO.
|
| 127 |
+
2. Saves results directly to `raw_data/FusionPDB_level2_fusion_structure_links.csv` and `raw_data/FusionPDB_level3_fusion_structure_links.csv`, respectively
|
| 128 |
+
3. Retrieves FO head gene and tail gene info
|
| 129 |
+
1. Using the tables collected in step (i), visits the page for each fusion oncoprotein (FO) in FusionPDB Level 2 and 3 to download head/tail info. Collects HGID and TGID (GeneIDs for head and tail) and UniProt accessions for each.
|
| 130 |
+
2. Saves results directly to `raw_data/level2_head_tail_info.txt` and `raw_data/level3_head_tail_info.txt`, respectively.
|
| 131 |
+
4. Combines Level 2 and 3 head/tail data
|
| 132 |
+
1. Merges `raw_data/level2_head_tail_info.txt` and `raw_data/level3_head_tail_info.txt` into a dataframe.
|
| 133 |
+
2. Saves result at `processed_data/fusionpdb/fusion_heads_and_tails.csv` (columns="FusionGID","HGID","TGID","HGUniProtAcc","TGUniProtAcc")
|
| 134 |
+
5. Combines Level 2 and 3 structure link data
|
| 135 |
+
1. Joins structure link data with metadata for each of levels 2 and 3, then combines the result.
|
| 136 |
+
2. Saves result at `processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_structure_links.csv`
|
| 137 |
+
6. Combines structure link data and metadata (result of step (v)) with head and tail data (result of step (iv)), and resolves any missing head/tail UniProt IDs.
|
| 138 |
+
1. Merges the data
|
| 139 |
+
2. Checks how many rows have either missing or wrong UniProt accessions for the head or tail gene, and compiles the gene symbols for online quering in the UniProt ID Mapping tool (`processed_data/fusionpdb/intermediates/unmapped_parts.txt`)
|
| 140 |
+
3. Reads the UniProt ID Mapping result. Combines this data with FusionPDB-scraped data by matching FusionPDB's HGID (GeneID for head) and TGID (GeneID for tail) with the GeneID returned by UniProt.
|
| 141 |
+
4. For any FO where FusionPDB lacked a UniProt ID for the head/tail, this ID is filled in from the UniProt ID Mapping result.
|
| 142 |
+
5. Saves result to `processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_head_tail_info.csv`. Columns: "FusionGID","FusionGene","Hgene","Tgene","URL","HGID","TGID","HGUniProtAcc","TGUniProtAcc","HGUniProtAcc_Source","TGUniProtAcc_Source", where the "_Source" columns indicate whether the UniProt ID came from FusionPDB, or from the ID Map.
|
| 143 |
+
7. Downloads AlphaFold2 structures of FOs from FusionPDB.
|
| 144 |
+
1. Using structure links from `processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_structure_links.csv` (step (v)), directly downloads `.pdb` and `.cif` files.
|
| 145 |
+
2. Saves results in 📁`raw_data/fusionpdb/structures`
|
| 146 |
+
|
| 147 |
+
<br>
|
| 148 |
+
|
| 149 |
+
2. 🐍 **`process_fusion_structures.py`**
|
| 150 |
+
1. Determines pLDDT(s) for each FO structure.
|
| 151 |
+
1. For each structure in 📁`raw_data/fusionpdb_structures/`, determines amino acid sequence, per-residue pLDDT, and average pLDDT from the AlphaFold2 structure.
|
| 152 |
+
2. Saves results in `processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_structures_processed.csv`.
|
| 153 |
+
2. Downloads AlphaFold2 structures for all head and tail proteins
|
| 154 |
+
1. Reads `processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_head_tail_info.csv` and collects all unique UniProt IDs for all head/tail proteins.
|
| 155 |
+
2. For each UniProt ID, queries the AlphaFoldDB, downloads the AlphaFold2 structure (if available), and saves it to 📁`raw_data/fusionpdb/head_tail_af2db_structures/`. Saves files converted from PDB to CIF format in `mmcif_converted_files`. Then, extracts the sequence, per-residue pLDDT, and average pLDDT from the file.
|
| 156 |
+
3. Saves any UniProt IDs that did not have structures in the AlphaFoldDB to: `processed_data/fusionpdb/intermediates/uniprotids_not_in_afdb.txt`. Most of these were very long, but the shorter ones were folded and their average pLDDTs were manually inputted. These were put back into the AlphaFold ID map to look for alternative UniProt IDs, and their results are in `not_in_afdb_idmap.txt`.
|
| 157 |
+
4. Saves results to `processed_data/fusionpdb/heads_tails_structural_data.csv`
|
| 158 |
+
3. Cleans the dataase of level 2&3 structural info
|
| 159 |
+
1. Drops rows where no structure was successfully downloaded
|
| 160 |
+
2. Drops rows where the FO sequence from FusionPDB does not match the FO sequence from its own AlphaFold2 structure file
|
| 161 |
+
3. ⭐️Saves **two final, cleaned databases**⭐️:
|
| 162 |
+
1. ⭐️ **`FusionPDB_level2-3_cleaned_FusionGID_info.csv`**: includes ful IDs and structural information for the Hgene and Tgene of each FO. Columns="FusionGID","FusionGene","Hgene","Tgene","URL","HGID","TGID","HGUniProtAcc","TGUniProtAcc","HGUniProtAcc_Source","TGUniProtAcc_Source","HG_pLDDT","HG_AA_pLDDTs","HG_Seq","TG_pLDDT","TG_AA_pLDDTs","TG_Seq".
|
| 163 |
+
2. ⭐️ **`FusionPDB_level2-3_cleaned_structure_info.csv`**: includes full structural information for each FO. Columns= "FusionGID","FusionGene","Fusion_Seq","Fusion_Length","Hgene","Hchr","Hbp","Hstrand","Tgene","Tchr","Tbp","Tstrand","Level","Fusion_Structure_Link","Fusion_Structure_Type","Fusion_pLDDT","Fusion_AA_pLDDTs","Fusion_Seq_Source"
|
| 164 |
+
|
| 165 |
+
|
| 166 |
+
### Training
|
| 167 |
+
|
| 168 |
+
The model is defined in `model.py` and `utils.py`. Training configs can be provided in `config.py`:
|
| 169 |
+
|
| 170 |
+
```
|
| 171 |
+
# Which models to benchmark
|
| 172 |
+
BENCHMARK_FUSONPLM = True
|
| 173 |
+
# FUSONPLM_CKPTS. If you've traiend your own model, this is a dictionary: key = run name, values = epochs
|
| 174 |
+
# If you want to use the trained FusOn-pLM, instead FUSONPLM_CKPTS="FusOn-pLM"
|
| 175 |
+
FUSONPLM_CKPTS= "FusOn-pLM"
|
| 176 |
+
|
| 177 |
+
BENCHMARK_ESM = True
|
| 178 |
+
|
| 179 |
+
# GPU configs
|
| 180 |
+
CUDA_VISIBLE_DEVICES="0"
|
| 181 |
+
|
| 182 |
+
# Overwriting configs
|
| 183 |
+
PERMISSION_TO_OVERWRITE_EMBEDDINGS = False # if False, script will halt if it believes these embeddings have already been made.
|
| 184 |
+
PERMISSION_TO_OVERWRITE_MODELS = False # if False, script will halt if it believes these embeddings have already been made.
|
| 185 |
+
```
|
| 186 |
+
|
| 187 |
+
<br>
|
| 188 |
+
|
| 189 |
+
`train.py` trains the models using embeddings indicated in `config.py`. It also performs a hyperparameter screen. Model raw outputs (probabilities) and performance metrics are saved in `trained_models`. For example, FusOn-pLM-Diso raw outputs (ESM-2-650M-Diso has a folder in the same format, and future trained models will as well):
|
| 190 |
+
|
| 191 |
+
```
|
| 192 |
+
benchmarking/
|
| 193 |
+
└── caid/
|
| 194 |
+
└── trained_models/
|
| 195 |
+
└── esm2_t33_650M_UR50D/best/
|
| 196 |
+
└── fuson_plm/best/
|
| 197 |
+
├── caid_hyperparam_screen_fusion_benchmark_metrics.csv
|
| 198 |
+
├── caid_hyperparam_screen_fusion_benchmark_probs.csv
|
| 199 |
+
├── caid_hyperparam_screen_test_metrics.csv
|
| 200 |
+
├── caid_hyperparam_screen_test_probs.csv
|
| 201 |
+
├── caid_train_losses.csv
|
| 202 |
+
├── params.txt
|
| 203 |
+
```
|
| 204 |
+
|
| 205 |
+
- **`caid_hyperparam_screen_fusion_benchmark_metrics.csv`**: performance metrics (Accuracy, Precision, Recall, F1 Score, AUROC) for the top model on the fusion benchmark set (`splits/fusion_bench_df.csv`)
|
| 206 |
+
- **`caid_hyperparam_screen_fusion_benchmark_probs.csv`**: for the fusion benchmark, raw probabilities of class 1 (disorder), threshold used to assign 0/1 based on maximized F1 score, prediction labels based on probabilities and threshold
|
| 207 |
+
- **`caid_hyperparam_screen_test_metrics.csv`**: same as `caid_hyperparam_screen_fusion_benchmark_metrics.csv`, but for CAID2 Disorder-NOX (`splits/test_df.csv`)
|
| 208 |
+
- **`caid_hyperparam_screen_test_probs.csv`**: same as `caid_hyperparam_screen_fusion_benchmark_probs`, but for CAID2 Disorder-NOX
|
| 209 |
+
- **`caid_train_losses.csv`**: train losses over the 2 training epochs for top-performing model
|
| 210 |
+
- **`params.txt`**: hyperparameters of top performing model
|
| 211 |
+
|
| 212 |
+
The training script also populates the `results` directory. Results from the FusOn-pLM manuscript are found in `results/final`. A few extra data files and plots are added by `analyze_fus`
|
| 213 |
+
|
| 214 |
+
```
|
| 215 |
+
benchmarking/
|
| 216 |
+
└── caid/
|
| 217 |
+
└── results/final
|
| 218 |
+
├── best_caid_model_results.csv
|
| 219 |
+
├── caid_hyperparam_screen_test_metrics.csv
|
| 220 |
+
├── caid_hyperparam_screen_fusion_benchmark_metrics.csv
|
| 221 |
+
├── caid_hyperparam_screen_train_losses.csv
|
| 222 |
+
├── fusion_disorder_boxplots.png
|
| 223 |
+
├── fusion_pred_disorder_r2.png
|
| 224 |
+
├── fusion_disorder_boxplots_source_data.csv
|
| 225 |
+
├── fusion_pred_disorder_r2_source_data.csv
|
| 226 |
+
├── CAID2_FusOn-pLM-Diso_with_ESM_AUROC_curve.png
|
| 227 |
+
├── CAID_fpr_tpr_source_data.csv
|
| 228 |
+
├── CAID_prediction_source_data.csv
|
| 229 |
+
```
|
| 230 |
+
|
| 231 |
+
- **`best_caid_model_results.csv`**: Summary file of hyperparameters, test set statistics, and fusion benchmark statistics for the best model of each type screened (ESM-2-650M, FusOn-pLM)
|
| 232 |
+
- **`caid_hyperparam_screen_fusion_benchmark_metrics.csv`**: Fusion benchmark set statistics for full hyperparameter screen
|
| 233 |
+
- **`caid_hyperparam_screen_fusion_benchmark_metrics.csv`**: Test set statistics for full hyperparameter screen
|
| 234 |
+
- **`caid_hyperparam_screen_train_losses.csv`**: Train losses for full hyperparameter screen
|
| 235 |
+
- 📊 **`fusion_disorder_boxplots.png`**: Fig. 4E, left (data directly used to produce the plot at `fusion_disorder_boxplots_source_data.csv`)
|
| 236 |
+
- 📊 **`fusion_pred_disorder_r2_source_data.csv`**: Fig. 4E, right (data directly used to produce the plot at `fusion_pred_disorder_r2_source_data.csv`)
|
| 237 |
+
- 📊 **`CAID2_FusOn-pLM-Diso_with_ESM_AUROC_curve.png`**: Fig. 4D (probabilities used at `CAID_prediction_source_data.csv`, FPR/TPR relationships directly used to make the plot at `CAID_fpr_tpr_source_data.csv`)
|
| 238 |
+
|
| 239 |
+
To run the training script, use
|
| 240 |
+
|
| 241 |
+
```
|
| 242 |
+
nohup python train.py > train.out 2> train.err &
|
| 243 |
+
```
|
| 244 |
+
|
| 245 |
+
### Plotting
|
| 246 |
+
|
| 247 |
+
The `plot.py` script generates many figures from the paper, alongside the formatted data directly used for plotting.
|
| 248 |
+
|
| 249 |
+
```
|
| 250 |
+
benchmarking/
|
| 251 |
+
└── caid/
|
| 252 |
+
└── results/final/
|
| 253 |
+
├── CAID2_FusOn-pLM-Diso_with_ESM_AUROC_curve.png
|
| 254 |
+
└── processed_data/
|
| 255 |
+
└── figures/
|
| 256 |
+
└── fusion_disorder/
|
| 257 |
+
├── plddt_sequence_EML4-ALK.png
|
| 258 |
+
├── plddt_sequence_EML4::ALK_source_data.csv
|
| 259 |
+
├── plddt_sequence_EWSR1-FLI1.png
|
| 260 |
+
├── plddt_sequence_EWSR1::FLI1_source_data.csv
|
| 261 |
+
├── plddt_sequence_PAX3-FOXO1.png
|
| 262 |
+
├── plddt_sequence_PAX3::FOXO1_source_data.csv
|
| 263 |
+
├── plddt_sequence_SS18-SSX1.png
|
| 264 |
+
├── plddt_sequence_SS18::SSX1_source_data.csv
|
| 265 |
+
└── histograms/
|
| 266 |
+
├── disorder_nox_histogram.png
|
| 267 |
+
├── disorder_nox_histogram_source_data.csv
|
| 268 |
+
├── fusions_histogram.png
|
| 269 |
+
├── fusions_histogram_source_data.csv
|
| 270 |
+
├── heads_histogram.png
|
| 271 |
+
├── heads_histogram_source_data.csv
|
| 272 |
+
├── tails_histogram.png
|
| 273 |
+
├── tails_histogram_source_data.csv
|
| 274 |
+
```
|
| 275 |
+
|
| 276 |
+
- Plots in `fusion_disorder` are from Fig. 1C
|
| 277 |
+
- Plots in `hisograms` are from Fig. 1D and Fig. S1
|
| 278 |
+
|
| 279 |
+
To regenerate these plots and source data, run:
|
| 280 |
+
|
| 281 |
+
```
|
| 282 |
+
python plot.py
|
| 283 |
+
```
|
| 284 |
+
|
| 285 |
+
### Colored structure images
|
| 286 |
+
`color_disorder_residues.ipynb` is used to plot fusion structures with pLDDT or disorder prediction color overlays. By running certain (or all) of its cells, you will recreate images from Fig. 1C and 4F, as well as the following file:
|
| 287 |
+
|
| 288 |
+
```
|
| 289 |
+
benchmarking/
|
| 290 |
+
└── caid/
|
| 291 |
+
└── disorder_coloring_data
|
| 292 |
+
├── normalized_disorder_propensities_source_data.csv
|
| 293 |
+
```
|
| 294 |
+
- **`normalized_disorder_propensities_source_data.csv`**: the normalized disorder propensities that were visualized on fusion structures in Fig. 4F
|
fuson_plm/benchmarking/caid/__init__.py
ADDED
|
File without changes
|
fuson_plm/benchmarking/caid/analyze_fusion_preds.py
ADDED
|
@@ -0,0 +1,158 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
|
| 2 |
+
import torch
|
| 3 |
+
import torch.nn as nn
|
| 4 |
+
import os
|
| 5 |
+
import pickle
|
| 6 |
+
import pandas as pd
|
| 7 |
+
import numpy as np
|
| 8 |
+
|
| 9 |
+
from sklearn.metrics import accuracy_score, precision_score, recall_score, f1_score, roc_auc_score, precision_recall_curve, average_precision_score
|
| 10 |
+
from fuson_plm.utils.logging import log_update, open_logfile
|
| 11 |
+
from fuson_plm.benchmarking.caid.plot import plot_fusion_stats_boxplots, plot_fusion_frac_disorder_r2
|
| 12 |
+
|
| 13 |
+
# calculate AUROC and AUPRC for each sequence
|
| 14 |
+
def calc_metrics(row):
|
| 15 |
+
probs = row['prob_1']
|
| 16 |
+
probs = [float(y) for y in probs.split(',')]
|
| 17 |
+
true_labels = row['Label']
|
| 18 |
+
true_labels = [int(y) for y in list(true_labels)]
|
| 19 |
+
pred_labels = row['pred_labels']
|
| 20 |
+
pred_labels = [int(y) for y in list(pred_labels)]
|
| 21 |
+
|
| 22 |
+
# Calculate AUROC
|
| 23 |
+
# Calculate AUPRC
|
| 24 |
+
# Calculate all the other stats based on the predicted labels
|
| 25 |
+
|
| 26 |
+
flat_binary_preds = np.array(pred_labels)
|
| 27 |
+
flat_prob_preds = np.array(probs)
|
| 28 |
+
flat_labels = np.array(true_labels)
|
| 29 |
+
|
| 30 |
+
accuracy = accuracy_score(flat_labels, flat_binary_preds)
|
| 31 |
+
precision = precision_score(flat_labels, flat_binary_preds)
|
| 32 |
+
recall = recall_score(flat_labels, flat_binary_preds)
|
| 33 |
+
f1 = f1_score(flat_labels, flat_binary_preds)
|
| 34 |
+
try:
|
| 35 |
+
roc_auc = roc_auc_score(flat_labels, flat_prob_preds)
|
| 36 |
+
except:
|
| 37 |
+
roc_auc = np.nan
|
| 38 |
+
|
| 39 |
+
try:
|
| 40 |
+
auprc = average_precision_score(flat_labels, flat_prob_preds)
|
| 41 |
+
except:
|
| 42 |
+
auprc = np.nan
|
| 43 |
+
|
| 44 |
+
return pd.Series({
|
| 45 |
+
'Accuracy': round(accuracy,3),
|
| 46 |
+
'Precision': round(precision,3),
|
| 47 |
+
'Recall': round(recall,3),
|
| 48 |
+
'F1': round(f1,3),
|
| 49 |
+
'AUROC': round(roc_auc,3) if not(np.isnan(roc_auc)) else roc_auc,
|
| 50 |
+
'AUPRC': round(auprc,3) if not(np.isnan(auprc)) else auprc,
|
| 51 |
+
})
|
| 52 |
+
|
| 53 |
+
def get_model_preds_with_metrics(path_to_model_predictions):
|
| 54 |
+
# Define paths and dataframes that we will need
|
| 55 |
+
fusion_benchmark_set = pd.read_csv('splits/fusion_bench_df.csv')
|
| 56 |
+
model_predictions = pd.read_csv(path_to_model_predictions)
|
| 57 |
+
fusion_structure_data = pd.read_csv('processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv')
|
| 58 |
+
fusion_structure_data['Fusion_Structure_Link'] = fusion_structure_data['Fusion_Structure_Link'].apply(lambda x: x.split('/')[-1])
|
| 59 |
+
|
| 60 |
+
# merge fusion data with seq ids
|
| 61 |
+
fuson_db = pd.read_csv('../../data/fuson_db.csv')
|
| 62 |
+
fuson_db = fuson_db[['aa_seq','seq_id']].rename(columns={'aa_seq':'Fusion_Seq'})
|
| 63 |
+
fusion_structure_data = pd.merge(
|
| 64 |
+
fusion_structure_data,
|
| 65 |
+
fuson_db,
|
| 66 |
+
on='Fusion_Seq',
|
| 67 |
+
how='inner'
|
| 68 |
+
)
|
| 69 |
+
|
| 70 |
+
# merge fusion structure data with top swissprot alignments
|
| 71 |
+
swissprot_top_alignments = pd.read_csv("../../data/blast/blast_outputs/swissprot_top_alignments.csv")
|
| 72 |
+
fusion_structure_data = pd.merge(
|
| 73 |
+
fusion_structure_data,
|
| 74 |
+
swissprot_top_alignments,
|
| 75 |
+
on="seq_id",
|
| 76 |
+
how="left"
|
| 77 |
+
)
|
| 78 |
+
|
| 79 |
+
model_predictions_labeled = pd.merge(model_predictions,fusion_benchmark_set.rename(columns={'Sequence':'sequence'}),on='sequence',how='inner')
|
| 80 |
+
model_predictions_labeled = pd.merge(model_predictions_labeled,
|
| 81 |
+
fusion_structure_data[['FusionGene','Fusion_Seq','Fusion_Structure_Link','Fusion_pLDDT','Fusion_AA_pLDDTs',
|
| 82 |
+
'top_hg_UniProtID', 'top_hg_UniProt_isoform', 'top_hg_UniProt_fus_indices', 'top_tg_UniProtID', 'top_tg_UniProt_isoform',
|
| 83 |
+
'top_tg_UniProt_fus_indices', 'top_UniProtID', 'top_UniProt_isoform', 'top_UniProt_fus_indices', 'top_UniProt_nIdentities',
|
| 84 |
+
'top_UniProt_nPositives']].rename(
|
| 85 |
+
columns={'Fusion_Seq': 'sequence'}
|
| 86 |
+
),
|
| 87 |
+
on='sequence',
|
| 88 |
+
how='left')
|
| 89 |
+
model_predictions_labeled['length'] = model_predictions_labeled['sequence'].str.len()
|
| 90 |
+
model_predictions_labeled['Fusion_Structure_Link'] = model_predictions_labeled['Fusion_Structure_Link'].apply(lambda x: x.split('/')[-1])
|
| 91 |
+
|
| 92 |
+
model_predictions_labeled[['Accuracy','Precision','Recall','F1','AUROC','AUPRC']] = model_predictions_labeled.apply(lambda row: calc_metrics(row),axis=1)
|
| 93 |
+
model_predictions_labeled = model_predictions_labeled.sort_values(by=['AUROC','F1','AUPRC','Accuracy','Precision','Recall'],ascending=[False,False,False,False,False,False]).reset_index(drop=True)
|
| 94 |
+
model_predictions_labeled['pcnt_disordered'] = round(100*model_predictions_labeled['Label'].apply(lambda x: sum([int(y) for y in list(x)]))/model_predictions_labeled['sequence'].str.len(),2)
|
| 95 |
+
model_predictions_labeled['pred_pcnt_disordered'] = round(100*model_predictions_labeled['pred_labels'].apply(lambda x: sum([int(y) for y in list(x)]))/model_predictions_labeled['sequence'].str.len(),2)
|
| 96 |
+
log_update(f"Model predictions for {len(model_predictions_labeled)} fusion oncoproteins. Preview:")
|
| 97 |
+
log_update(
|
| 98 |
+
model_predictions_labeled[['sequence','length','FusionGene','Fusion_pLDDT','pcnt_disordered','pred_pcnt_disordered','AUROC','F1','AUPRC','Accuracy','Precision','Recall']].head()
|
| 99 |
+
)
|
| 100 |
+
cols_str = '\n\t'+ '\n\t'.join(list(model_predictions_labeled.columns))
|
| 101 |
+
log_update(f"Columns in model predictions stats database: {cols_str}")
|
| 102 |
+
|
| 103 |
+
# There is one duplicate row
|
| 104 |
+
duplicate_sequences = model_predictions_labeled.loc[model_predictions_labeled['sequence'].duplicated()]['sequence'].unique().tolist()
|
| 105 |
+
log_update(f"\nTotal duplicate sequences: {len(duplicate_sequences)}")
|
| 106 |
+
gb = model_predictions_labeled.groupby('sequence').agg(
|
| 107 |
+
pred_labels=("pred_labels", list),
|
| 108 |
+
)
|
| 109 |
+
gb['pred_labels'] = gb['pred_labels'].apply(lambda x: list(set(x)))
|
| 110 |
+
gb['unique_label_vectors'] = gb['pred_labels'].apply(lambda x: len(x))
|
| 111 |
+
log_update(f"Duplicate entries for sequences have the exact same label vector: {(gb['unique_label_vectors']==1).all()}")
|
| 112 |
+
log_update("\tSince above statement is true, randomly dropping duplicate sequence rows - should make no difference to prediction.")
|
| 113 |
+
|
| 114 |
+
model_predictions_labeled = model_predictions_labeled.drop_duplicates('sequence').reset_index(drop=True)
|
| 115 |
+
#os.makedirs("processed_data/fusion_predictions",exist_ok=True)
|
| 116 |
+
return model_predictions_labeled
|
| 117 |
+
|
| 118 |
+
def calc_average_stats(model_pred_stats):
|
| 119 |
+
# cols: Accuracy Precision Recall F1 AUROC AUPRC pcnt_disordered pred_pcnt_disordered
|
| 120 |
+
averages = model_pred_stats[[
|
| 121 |
+
'Accuracy', 'Precision', 'Recall', 'F1', 'AUROC', 'AUPRC'
|
| 122 |
+
]].mean()
|
| 123 |
+
averages
|
| 124 |
+
|
| 125 |
+
def main():
|
| 126 |
+
with open_logfile("analyze_fusion_preds.txt"):
|
| 127 |
+
## Put path to model predictions you'd like to use for benchmarking
|
| 128 |
+
path_to_model_predictions = "trained_models/fuson_plm/best/caid_hyperparam_screen_fusion_benchmark_probs.csv"
|
| 129 |
+
save_dir = "results/final"
|
| 130 |
+
preds_with_metrics_save_path = f"{save_dir}/model_preds_with_metrics.csv"
|
| 131 |
+
boxplot_save_path = f"{save_dir}/fusion_disorder_boxplots.png"
|
| 132 |
+
r2_save_path = "results/final/fusion_pred_disorder_r2.png"
|
| 133 |
+
|
| 134 |
+
# Additional benchmarking on fusion predictions
|
| 135 |
+
fuson_db = pd.read_csv("../../data/fuson_db.csv")
|
| 136 |
+
seq_id_dict = dict(zip(fuson_db['aa_seq'],fuson_db['seq_id']))
|
| 137 |
+
model_preds_with_metrics = get_model_preds_with_metrics(path_to_model_predictions)
|
| 138 |
+
model_preds_with_metrics['seq_id'] = model_preds_with_metrics['sequence'].map(seq_id_dict)
|
| 139 |
+
model_preds_with_metrics.to_csv(preds_with_metrics_save_path,index=False)
|
| 140 |
+
print(model_preds_with_metrics.columns)
|
| 141 |
+
|
| 142 |
+
# Box plot
|
| 143 |
+
boxplot_model_preds = model_preds_with_metrics[['seq_id','FusionGene',
|
| 144 |
+
'Accuracy', 'Precision', 'Recall', 'F1', 'AUROC'
|
| 145 |
+
]]
|
| 146 |
+
|
| 147 |
+
boxplot_model_preds.to_csv(boxplot_save_path.replace(".png","_source_data.csv"),index=False)
|
| 148 |
+
plot_fusion_stats_boxplots(boxplot_model_preds[['Accuracy', 'Precision', 'Recall', 'F1', 'AUROC'
|
| 149 |
+
]], save_path=boxplot_save_path)
|
| 150 |
+
|
| 151 |
+
# R2 plot
|
| 152 |
+
r2_model_preds = model_preds_with_metrics[['seq_id','FusionGene','pcnt_disordered','pred_pcnt_disordered']]
|
| 153 |
+
r2_model_preds.to_csv(r2_save_path.replace(".png","_source_data.csv"),index=False)
|
| 154 |
+
plot_fusion_frac_disorder_r2(r2_model_preds['pcnt_disordered'], r2_model_preds['pred_pcnt_disordered'], save_path=r2_save_path)
|
| 155 |
+
calc_average_stats(model_preds_with_metrics)
|
| 156 |
+
|
| 157 |
+
if __name__ == "__main__":
|
| 158 |
+
main()
|
fuson_plm/benchmarking/caid/clean.py
ADDED
|
@@ -0,0 +1,671 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
import os
|
| 2 |
+
import numpy as np
|
| 3 |
+
import re
|
| 4 |
+
import pandas as pd
|
| 5 |
+
import requests
|
| 6 |
+
|
| 7 |
+
from fuson_plm.utils.logging import open_logfile, log_update
|
| 8 |
+
from fuson_plm.utils.constants import DELIMITERS, VALID_AAS
|
| 9 |
+
from fuson_plm.utils.data_cleaning import check_columns_for_listlike, find_invalid_chars
|
| 10 |
+
|
| 11 |
+
from fuson_plm.benchmarking.caid.scrape_fusionpdb import scrape_fusionpdb_level_2_3
|
| 12 |
+
from fuson_plm.benchmarking.caid.process_fusion_structures import process_fusions_and_hts
|
| 13 |
+
|
| 14 |
+
def download_fasta(uniprotid, includeIsoform, output_file):
|
| 15 |
+
try:
|
| 16 |
+
url = f"https://rest.uniprot.org/uniprotkb/search?format=fasta&includeIsoform={includeIsoform}&query=accession%3A{uniprotid}&size=500&sort=accession+asc"
|
| 17 |
+
# Send a GET request to the URL
|
| 18 |
+
response = requests.get(url)
|
| 19 |
+
|
| 20 |
+
# Raise an exception if the request was unsuccessful
|
| 21 |
+
response.raise_for_status()
|
| 22 |
+
|
| 23 |
+
# Write the content to a file in text mode
|
| 24 |
+
with open(output_file, 'a+') as file:
|
| 25 |
+
file.write(response.text)
|
| 26 |
+
|
| 27 |
+
log_update(f"FASTA file for {uniprotid} successfully downloaded and added to '{output_file}'")
|
| 28 |
+
|
| 29 |
+
except requests.exceptions.RequestException as e:
|
| 30 |
+
log_update(f"An error occurred: {e}")
|
| 31 |
+
|
| 32 |
+
# Test Sequences (CAID-2 Disorder-NOX)
|
| 33 |
+
def parse_caid_txt(fast_file):
|
| 34 |
+
'''
|
| 35 |
+
Parses correctly fasta-formatted text file with conditions:
|
| 36 |
+
Line 1: ID
|
| 37 |
+
Line 2: Sequence
|
| 38 |
+
Line 3: Label
|
| 39 |
+
'''
|
| 40 |
+
|
| 41 |
+
seq_to_label = {}
|
| 42 |
+
id_to_sequence = {}
|
| 43 |
+
|
| 44 |
+
with open(fast_file, 'r') as file:
|
| 45 |
+
label = None
|
| 46 |
+
sequence = ""
|
| 47 |
+
seq_id = None
|
| 48 |
+
reading_sequence = False
|
| 49 |
+
for line in file:
|
| 50 |
+
line = line.strip()
|
| 51 |
+
if line.startswith(">"):
|
| 52 |
+
if label is not None and sequence:
|
| 53 |
+
seq_to_label[sequence] = (label, seq_id)
|
| 54 |
+
seq_id = line[1:] # Capture the ID without the '>'
|
| 55 |
+
label = None
|
| 56 |
+
sequence = ""
|
| 57 |
+
reading_sequence = True
|
| 58 |
+
elif reading_sequence:
|
| 59 |
+
if all(c in "01-" for c in line):
|
| 60 |
+
label = line
|
| 61 |
+
reading_sequence = False
|
| 62 |
+
else:
|
| 63 |
+
sequence += line
|
| 64 |
+
if label is not None and sequence:
|
| 65 |
+
seq_to_label[sequence] = (label, seq_id)
|
| 66 |
+
|
| 67 |
+
return seq_to_label
|
| 68 |
+
|
| 69 |
+
def check_df_for_mismatched_labels(sd):
|
| 70 |
+
log_update("\tChecking dataframe for mismatched sequences and labels...")
|
| 71 |
+
counter=0
|
| 72 |
+
for idx, row in sd.iterrows():
|
| 73 |
+
seq = row['Sequence']
|
| 74 |
+
label = row['Label']
|
| 75 |
+
|
| 76 |
+
if len(seq) != len(label):
|
| 77 |
+
counter+=1
|
| 78 |
+
log_update(f"\t\tLength mismatch at index {idx}: sequence length = {len(seq)}, label length = {len(label)}")
|
| 79 |
+
|
| 80 |
+
log_update(f"\t\tTotal mismatched lengths/labels: {counter}")
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
def process_caid2_disorder_nox_test(caid_path):
|
| 84 |
+
"""
|
| 85 |
+
Processes the CAID-2_Disorder_NOX_Testing_Sequences.fasta file
|
| 86 |
+
"""
|
| 87 |
+
log_update("Processing CAID-2-Disorder-NOX Testing Dataset")
|
| 88 |
+
# Parse the fasta file
|
| 89 |
+
caid_dict = parse_caid_txt(caid_path)
|
| 90 |
+
|
| 91 |
+
# Gather the sequences
|
| 92 |
+
caid_seqs = {}
|
| 93 |
+
for k, (v, seq_id) in caid_dict.items():
|
| 94 |
+
caid_seqs[seq_id] = (k, v)
|
| 95 |
+
log_update(f"\tTotal sequences: {len(caid_seqs)}")
|
| 96 |
+
|
| 97 |
+
# Form dataframe from processed data
|
| 98 |
+
caid_df = pd.DataFrame({
|
| 99 |
+
'ID': list(caid_seqs.keys()),
|
| 100 |
+
'Sequence': [seq for seq, _ in caid_seqs.values()],
|
| 101 |
+
'Label': [lbl for _, lbl in caid_seqs.values()],
|
| 102 |
+
'Split': 'Test'
|
| 103 |
+
})
|
| 104 |
+
|
| 105 |
+
check_df_for_mismatched_labels(caid_df)
|
| 106 |
+
return caid_df
|
| 107 |
+
|
| 108 |
+
# Training Squences (fldpnn and IDP-CRF)
|
| 109 |
+
# fldpnn Training Sequences
|
| 110 |
+
def parse_fldpnn_fasta(file_path):
|
| 111 |
+
"""
|
| 112 |
+
Parse flDPnn_Training_Dataset.txt, where there are 5 sequence lines. We only want the first
|
| 113 |
+
|
| 114 |
+
>Disprot ID
|
| 115 |
+
Amino acid sequence
|
| 116 |
+
Experimental annotation for intrinsic disorder
|
| 117 |
+
Experimental annotation for disordered protein binding
|
| 118 |
+
Experimental annotation for disordered DNA binding
|
| 119 |
+
Experimental annotation for disordered RNA binding
|
| 120 |
+
Experimental annotation for disordered flexible linkers
|
| 121 |
+
"""
|
| 122 |
+
sequences = []
|
| 123 |
+
labels = []
|
| 124 |
+
ids = []
|
| 125 |
+
|
| 126 |
+
with open(file_path, 'r') as file:
|
| 127 |
+
lines = file.readlines()
|
| 128 |
+
|
| 129 |
+
seq_id = ""
|
| 130 |
+
current_sequence = ""
|
| 131 |
+
seen_label_lines = 0 # should go up to 5 for each
|
| 132 |
+
current_labels = []
|
| 133 |
+
is_label = False
|
| 134 |
+
|
| 135 |
+
for line in lines:
|
| 136 |
+
line = line.strip()
|
| 137 |
+
if line.startswith('>'):
|
| 138 |
+
if current_sequence and current_labels:
|
| 139 |
+
assert seen_label_lines==5 # we should've seen 5 labels, otherwise something is wrong
|
| 140 |
+
ids.append(seq_id)
|
| 141 |
+
sequences.append(current_sequence)
|
| 142 |
+
labels.append(''.join(current_labels))
|
| 143 |
+
seq_id = line[1:] # Capture the ID without the '>'
|
| 144 |
+
current_sequence = ""
|
| 145 |
+
current_labels = []
|
| 146 |
+
is_label = False
|
| 147 |
+
seen_label_lines = 0
|
| 148 |
+
elif re.match('^[A-Z]+$', line): # Sequence lines
|
| 149 |
+
current_sequence += line
|
| 150 |
+
else: # Label lines
|
| 151 |
+
seen_label_lines+=1
|
| 152 |
+
if seen_label_lines==1:
|
| 153 |
+
current_labels.append(line)
|
| 154 |
+
is_label = True
|
| 155 |
+
|
| 156 |
+
# Add the last sequence and labels
|
| 157 |
+
if current_sequence and current_labels:
|
| 158 |
+
sequences.append(current_sequence)
|
| 159 |
+
labels.append(''.join(current_labels))
|
| 160 |
+
ids.append(seq_id)
|
| 161 |
+
|
| 162 |
+
return ids, sequences, labels
|
| 163 |
+
|
| 164 |
+
def parse_idp_crf_fasta(file_path):
|
| 165 |
+
sequences = []
|
| 166 |
+
labels = []
|
| 167 |
+
ids = []
|
| 168 |
+
|
| 169 |
+
with open(file_path, 'r') as file:
|
| 170 |
+
lines = file.readlines()
|
| 171 |
+
|
| 172 |
+
seq_id = ""
|
| 173 |
+
current_sequence = ""
|
| 174 |
+
current_labels = []
|
| 175 |
+
is_label = False
|
| 176 |
+
|
| 177 |
+
for line in lines:
|
| 178 |
+
line = line.strip()
|
| 179 |
+
if line.startswith('>'):
|
| 180 |
+
if current_sequence and current_labels:
|
| 181 |
+
ids.append(seq_id)
|
| 182 |
+
sequences.append(current_sequence)
|
| 183 |
+
labels.append(''.join(current_labels))
|
| 184 |
+
seq_id = line[1:] # Capture the ID without the '>'
|
| 185 |
+
current_sequence = ""
|
| 186 |
+
current_labels = []
|
| 187 |
+
is_label = False
|
| 188 |
+
elif re.match('^[A-Z]+$', line): # Sequence lines
|
| 189 |
+
current_sequence += line
|
| 190 |
+
else: # Label lines
|
| 191 |
+
current_labels.append(line)
|
| 192 |
+
is_label = True
|
| 193 |
+
|
| 194 |
+
# Add the last sequence and labels
|
| 195 |
+
if current_sequence and current_labels:
|
| 196 |
+
sequences.append(current_sequence)
|
| 197 |
+
labels.append(''.join(current_labels))
|
| 198 |
+
ids.append(seq_id)
|
| 199 |
+
|
| 200 |
+
return ids, sequences, labels
|
| 201 |
+
|
| 202 |
+
def process_fldpnn(fldpnn_path, split="training"):
|
| 203 |
+
"""
|
| 204 |
+
Process the fldpnn_Training_Dataset
|
| 205 |
+
"""
|
| 206 |
+
log_update(f"\nProcessing flDPnn {split} dataset")
|
| 207 |
+
# Parse fasta
|
| 208 |
+
fldpnn_ids, fldpnn_seqs, fldpnn_labels = parse_fldpnn_fasta(fldpnn_path)
|
| 209 |
+
|
| 210 |
+
# Collect cleaned labels
|
| 211 |
+
cleaned_fldpnn_ids = []
|
| 212 |
+
cleaned_fldpnn_labels = []
|
| 213 |
+
for i in range(len(fldpnn_seqs)):
|
| 214 |
+
seq_len = len(fldpnn_seqs[i])
|
| 215 |
+
label = fldpnn_labels[i] # Should only be the first set of labels
|
| 216 |
+
id = fldpnn_ids[i]
|
| 217 |
+
cleaned_fldpnn_labels.append(label)
|
| 218 |
+
|
| 219 |
+
log_update(f"\tTotal labels: {len(cleaned_fldpnn_labels)}, total sequences: {len(fldpnn_seqs)},total IDs: {len(fldpnn_ids)}")
|
| 220 |
+
|
| 221 |
+
fldpnn_df = pd.DataFrame({'Sequence': fldpnn_seqs,
|
| 222 |
+
'Label': cleaned_fldpnn_labels,
|
| 223 |
+
"Split": "Train" if split=="training" else "Val",
|
| 224 |
+
"ID": fldpnn_ids})
|
| 225 |
+
check_df_for_mismatched_labels(fldpnn_df)
|
| 226 |
+
|
| 227 |
+
return fldpnn_df
|
| 228 |
+
|
| 229 |
+
def combine_fldpnn_train_val(fldpnn_train_df, fldpnn_val_df):
|
| 230 |
+
log_update("\nJoining flDPnn train and val sets into one training set for CAID predictor")
|
| 231 |
+
combined = pd.concat([fldpnn_train_df,fldpnn_val_df])
|
| 232 |
+
|
| 233 |
+
# check for duplicates
|
| 234 |
+
duplicates = combined[combined['Sequence'].duplicated()]['Sequence'].unique().tolist()
|
| 235 |
+
n_rows_with_duplicates = len(combined[combined['Sequence'].isin(duplicates)])
|
| 236 |
+
log_update(f"\t{len(duplicates)} sequences in both train and val datasets, corresponding to {n_rows_with_duplicates} rows")
|
| 237 |
+
for dup in duplicates:
|
| 238 |
+
train_id = combined.loc[(combined['Sequence']==dup) & (combined['Split']=='Train')]['ID'].reset_index(drop=True).iloc[0]
|
| 239 |
+
val_id = combined.loc[(combined['Sequence']==dup) & (combined['Split']=='Val')]['ID'].reset_index(drop=True).iloc[0]
|
| 240 |
+
train_label = combined.loc[(combined['Sequence']==dup) & (combined['Split']=='Train')]['Label'].reset_index(drop=True).iloc[0]
|
| 241 |
+
val_label = combined.loc[(combined['Sequence']==dup) & (combined['Split']=='Val')]['Label'].reset_index(drop=True).iloc[0]
|
| 242 |
+
log_update(f"\t\tTrain ID: {train_id}\tVal ID: {val_id}\tSame labels: {train_label==val_label}\tSequence: {dup}")
|
| 243 |
+
|
| 244 |
+
# if the labels are not equal, get rid of it completely. Otherwise just get rid of the val case
|
| 245 |
+
if not(train_label==val_label):
|
| 246 |
+
log_update(f"\t\t\tSince labels are not equal, removing sequence completely")
|
| 247 |
+
combined = combined[combined['Sequence']!=dup].reset_index(drop=True)
|
| 248 |
+
else:
|
| 249 |
+
log_update(f"\t\t\tSince labels are equal, removing validation copy")
|
| 250 |
+
combined = combined.loc[(combined['Sequence']!=dup) |
|
| 251 |
+
((combined['Sequence']==dup) & (combined['Split']=='Train'))]
|
| 252 |
+
# drop duplicates
|
| 253 |
+
log_update(f"\tLength of joined flDPnn data: {len(combined)}")
|
| 254 |
+
|
| 255 |
+
return combined
|
| 256 |
+
|
| 257 |
+
def process_idp_crf_train(idp_crf_train_path):
|
| 258 |
+
"""
|
| 259 |
+
Process IDP-CRF_Training_Dataset
|
| 260 |
+
|
| 261 |
+
Args:
|
| 262 |
+
idp_crf_train_path
|
| 263 |
+
"""
|
| 264 |
+
log_update("\nProcessing IDP-CRF training dataset")
|
| 265 |
+
# Parse the fasta, get sequences and labels
|
| 266 |
+
idp_crf_ids, idp_crf_seqs, idp_crf_labels = parse_idp_crf_fasta(idp_crf_train_path)
|
| 267 |
+
log_update(f"\tTotal labels: {len(idp_crf_labels)}, total sequences: {len(idp_crf_seqs)}, total IDs: {len(idp_crf_ids)}")
|
| 268 |
+
|
| 269 |
+
# Clean the labels
|
| 270 |
+
cleaned_idp_ids, cleaned_idp_seqs, cleaned_idp_labels = [], [], []
|
| 271 |
+
counter = 0
|
| 272 |
+
log_update("\tCleaning labels and counting length-mismatched examples...")
|
| 273 |
+
for i, label in enumerate(idp_crf_labels):
|
| 274 |
+
# If length of sequence and labels doesn't match, log it
|
| 275 |
+
if len(idp_crf_seqs[i]) != len(idp_crf_labels[i]):
|
| 276 |
+
log_update(f"\t\tLength mismatch at index {i}: sequence length = {len(idp_crf_seqs[i])}, label length = {len(idp_crf_labels[i])}")
|
| 277 |
+
|
| 278 |
+
counter += 1
|
| 279 |
+
# Else, "clean" the labels by mapping them to ints and converting them to a list
|
| 280 |
+
else:
|
| 281 |
+
cleaned_idp_ids.append(idp_crf_ids[i])
|
| 282 |
+
cleaned_idp_labels.append(label)
|
| 283 |
+
cleaned_idp_seqs.append(idp_crf_seqs[i])
|
| 284 |
+
|
| 285 |
+
log_update(f"\t\tMismatched lengths/labels: {counter}")
|
| 286 |
+
|
| 287 |
+
# Confirm that final database has no mismatched labels
|
| 288 |
+
idp_crf_df = pd.DataFrame({'Sequence': cleaned_idp_seqs,
|
| 289 |
+
'Label': cleaned_idp_labels,
|
| 290 |
+
"Split": "Train",
|
| 291 |
+
"ID": cleaned_idp_ids})
|
| 292 |
+
check_df_for_mismatched_labels(idp_crf_df)
|
| 293 |
+
|
| 294 |
+
return idp_crf_df
|
| 295 |
+
|
| 296 |
+
def find_agreeing_labels(row, lab1="", lab2=""):
|
| 297 |
+
"""
|
| 298 |
+
If there's only one possible label, return that label. If the two labels disagree, return np.nan
|
| 299 |
+
"""
|
| 300 |
+
val1 = row[lab1]
|
| 301 |
+
val2 = row[lab2]
|
| 302 |
+
|
| 303 |
+
# If one of them is nan, then they won't match anyway, so return True because there is no conflict
|
| 304 |
+
if type(val1)==float and np.isnan(val1):
|
| 305 |
+
return val2
|
| 306 |
+
elif type(val2)==float and np.isnan(val2):
|
| 307 |
+
return val1
|
| 308 |
+
else:
|
| 309 |
+
if val1==val2:
|
| 310 |
+
return val1
|
| 311 |
+
else:
|
| 312 |
+
return np.nan
|
| 313 |
+
|
| 314 |
+
def get_unique_ids(row):
|
| 315 |
+
source_to_id = {
|
| 316 |
+
"IDP-CRF": row["IDP-CRF ID"],
|
| 317 |
+
"flDPnn": row["flDPnn ID"],
|
| 318 |
+
"CAID-2_Disorder_NOX": row["CAID-2_Disorder_NOX ID"]
|
| 319 |
+
}
|
| 320 |
+
|
| 321 |
+
all_sources = row["Source"].split(",")
|
| 322 |
+
all_ids = []
|
| 323 |
+
# they are already in the desired order so just iterate through them
|
| 324 |
+
for source in all_sources:
|
| 325 |
+
candidate_id = source_to_id[source]
|
| 326 |
+
if not(candidate_id in all_ids):
|
| 327 |
+
all_ids.append(candidate_id)
|
| 328 |
+
|
| 329 |
+
return ",".join(all_ids)
|
| 330 |
+
|
| 331 |
+
def parse_caid2_results(processed_caid2_df,lines):
|
| 332 |
+
# iterate through the lines
|
| 333 |
+
all_caid2_disorder_nox_ids = processed_caid2_df['ID'].tolist()
|
| 334 |
+
all_caid2_disorder_nox_sequences = processed_caid2_df['Sequence'].tolist()
|
| 335 |
+
|
| 336 |
+
cur_id = None
|
| 337 |
+
results = {
|
| 338 |
+
}
|
| 339 |
+
for i, line in enumerate(lines):
|
| 340 |
+
# If line starts with >, that means we have a new ID
|
| 341 |
+
if line[0]==">":
|
| 342 |
+
# If we are currently on a different cur_id, finish that one out
|
| 343 |
+
if not(cur_id is None):
|
| 344 |
+
results[cur_id]['prob_1'] = ",".join(results[cur_id]['prob_1'])
|
| 345 |
+
results[cur_id]['pred_labels'] = ",".join(results[cur_id]['pred_labels'])
|
| 346 |
+
sequence = results[cur_id]['sequence']
|
| 347 |
+
# get the true labels from the CAID2 dataset - IF POSSIBLE
|
| 348 |
+
if (cur_id not in all_caid2_disorder_nox_ids) and (sequence not in all_caid2_disorder_nox_sequences):
|
| 349 |
+
results[cur_id]['labels'] = np.nan
|
| 350 |
+
else:
|
| 351 |
+
true_labels = processed_caid2_df.loc[
|
| 352 |
+
processed_caid2_df['ID']==cur_id,'Label'
|
| 353 |
+
].item()
|
| 354 |
+
true_labels = ",".join(list(true_labels))
|
| 355 |
+
results[cur_id]['labels'] = true_labels
|
| 356 |
+
# Now process the new one
|
| 357 |
+
cur_id = line[1::].strip('\t').strip('\n')
|
| 358 |
+
results[cur_id] = {
|
| 359 |
+
'sequence': '',
|
| 360 |
+
'prob_1': [],
|
| 361 |
+
'pred_labels': []
|
| 362 |
+
}
|
| 363 |
+
# if cur id is not None
|
| 364 |
+
else:
|
| 365 |
+
# if we have a cur id to process, process it
|
| 366 |
+
if not(cur_id is None):
|
| 367 |
+
# Extract the information - not every .caid file as predicted labels!!
|
| 368 |
+
lsplit = line.strip('\n').split('\t')
|
| 369 |
+
label=''
|
| 370 |
+
idx, aa, prob = lsplit[0], lsplit[1], lsplit[2]
|
| 371 |
+
if len(lsplit)==4: label=lsplit[3]
|
| 372 |
+
# Add to dict
|
| 373 |
+
results[cur_id]['sequence']+=aa
|
| 374 |
+
results[cur_id]['prob_1'].append(prob)
|
| 375 |
+
results[cur_id]['pred_labels'].append(label)
|
| 376 |
+
|
| 377 |
+
# if we're on the last line, combine
|
| 378 |
+
if i==len(lines)-1:
|
| 379 |
+
results[cur_id]['prob_1'] = ",".join(results[cur_id]['prob_1'])
|
| 380 |
+
results[cur_id]['pred_labels'] = ",".join(results[cur_id]['pred_labels'])
|
| 381 |
+
sequence = results[cur_id]['sequence']
|
| 382 |
+
# get the true labels from the CAID2 dataset - IF POSSIBLE
|
| 383 |
+
if (cur_id not in all_caid2_disorder_nox_ids) and (sequence not in all_caid2_disorder_nox_sequences):
|
| 384 |
+
results[cur_id]['labels'] = np.nan
|
| 385 |
+
else:
|
| 386 |
+
true_labels = processed_caid2_df.loc[
|
| 387 |
+
processed_caid2_df['ID']==cur_id,'Label'
|
| 388 |
+
].item()
|
| 389 |
+
true_labels = ",".join(list(true_labels))
|
| 390 |
+
results[cur_id]['labels'] = true_labels
|
| 391 |
+
|
| 392 |
+
df = pd.DataFrame.from_dict(results,orient='index').reset_index().rename(columns={'index':'seq_id'})
|
| 393 |
+
df = df.loc[df['labels'].notna()].reset_index(drop=True)
|
| 394 |
+
# drop pred_labels if it's empty
|
| 395 |
+
if set(','.join(df['pred_labels'].tolist()))=={','}:
|
| 396 |
+
df = df.drop(columns=['pred_labels'])
|
| 397 |
+
log_update(f"\t\tno predicted labels provided for this dataset; only probabilities")
|
| 398 |
+
log_update(f"\t\t{len(df)}/{len(all_caid2_disorder_nox_sequences)} total CAID2-Nox sequences")
|
| 399 |
+
return df
|
| 400 |
+
|
| 401 |
+
def parse_all_caid2_results(processed_caid2_df, caid_raw_folder="raw_data/caid2_competition_results"):
|
| 402 |
+
save_dir ="processed_data/caid2_competition_results"
|
| 403 |
+
os.makedirs(save_dir,exist_ok=True)
|
| 404 |
+
|
| 405 |
+
log_update(f"\nExtracting all CAID-2_Disorder_NOX results from CAID2 competition results files...")
|
| 406 |
+
all_caid_files = os.listdir(caid_raw_folder)
|
| 407 |
+
for caid_file in all_caid_files:
|
| 408 |
+
# figure out how to parse .caid files
|
| 409 |
+
with open(f"{caid_raw_folder}/{caid_file}", "r") as f:
|
| 410 |
+
lines = f.readlines()
|
| 411 |
+
log_update(f"\t{caid_file}:")
|
| 412 |
+
results_df = parse_caid2_results(processed_caid2_df,lines)
|
| 413 |
+
# save it
|
| 414 |
+
competitor_name = caid_file.split('.caid')[0]
|
| 415 |
+
results_df.to_csv(f"{save_dir}/{competitor_name}_CAID-2_Disorder_NOX.csv",index=False)
|
| 416 |
+
|
| 417 |
+
def make_train_df(fldpnn_df, idp_crf_df):
|
| 418 |
+
"""
|
| 419 |
+
Make training dataframe by concatenating the two processed training sets.
|
| 420 |
+
"""
|
| 421 |
+
# Add source columns so we can track where each sequence came from
|
| 422 |
+
idp_crf_df = idp_crf_df.rename(columns={'Label':'IDP-CRF Label', 'ID': 'IDP-CRF ID'}).drop(columns=['Split'])
|
| 423 |
+
fldpnn_df = fldpnn_df.rename(columns={'Label':'flDPnn Label', 'ID': 'flDPnn ID'}).drop(columns=['Split'])
|
| 424 |
+
########### Combine fldpnn and idp crf
|
| 425 |
+
# Join
|
| 426 |
+
log_update("\nJoining flDPnn and IDP-CRF data by sequence make unified training set")
|
| 427 |
+
train_df = pd.merge(idp_crf_df,
|
| 428 |
+
fldpnn_df,
|
| 429 |
+
on='Sequence',
|
| 430 |
+
how='outer',
|
| 431 |
+
indicator=True)
|
| 432 |
+
train_df["Split"] = ["Train"]*len(train_df)
|
| 433 |
+
# Map _merge column to desired labels
|
| 434 |
+
train_df['Source'] = train_df['_merge'].map({
|
| 435 |
+
'left_only': 'IDP-CRF',
|
| 436 |
+
'right_only': 'flDPnn',
|
| 437 |
+
'both': 'IDP-CRF,flDPnn'
|
| 438 |
+
})
|
| 439 |
+
train_df = train_df.drop(columns=["_merge"])
|
| 440 |
+
log_update(f"\tIDP-CRF dataset size: {len(idp_crf_df)}\n\tfLDpnn dataset size: {len(fldpnn_df)}\n\tinitial train dataset size: {len(train_df)}")
|
| 441 |
+
|
| 442 |
+
# Check for duplicate sequences
|
| 443 |
+
log_update(f"\tChecking for sequences in both datasets...")
|
| 444 |
+
duplicates = train_df[train_df["Source"].str.contains(",")]['Sequence'].unique().tolist()
|
| 445 |
+
n_rows_with_duplicates = len(train_df[train_df['Sequence'].isin(duplicates)])
|
| 446 |
+
log_update(f"\t\t{len(duplicates)} sequences in both datasets, corresponding to {n_rows_with_duplicates} rows")
|
| 447 |
+
|
| 448 |
+
# Check for consistency between IDP-CRF Label and flDPnn label
|
| 449 |
+
train_df["Label"] = train_df.apply(lambda row: find_agreeing_labels(row,lab1="IDP-CRF Label",lab2="flDPnn Label"),axis=1)
|
| 450 |
+
train_df["No Label Conflicts"]= ~train_df["Label"].isna()
|
| 451 |
+
log_update(f"\tChecked for label inconsistencies between IDP-CRF and flDPnn on the same sequence:")
|
| 452 |
+
match_str = train_df['No Label Conflicts'].value_counts().reset_index().rename(columns={'index': 'No Label Conflicts','No Label Conflicts': 'count'}).to_string(index=False)
|
| 453 |
+
match_str = "\t\t" + match_str.replace("\n","\n\t\t")
|
| 454 |
+
log_update(match_str)
|
| 455 |
+
|
| 456 |
+
# Dropping rows where labels don't match
|
| 457 |
+
#train_df[train_df['No Label Conflicts']==False][['Sequence','Split','IDP-CRF ID','flDPnn ID','IDP-CRF Label','flDPnn Label','No Label Conflicts']].to_csv('mismatch.csv',index=False)
|
| 458 |
+
# Drop row with known conflict with disprot
|
| 459 |
+
conflict_seq="MASREEEQRETTPERGRGAARRPPTMEDVSSPSPSPPPPRAPPKKRMRRRIESEDEEDSSQDALVPRTPSPRPSTSAADLAIAPKKKKKRPSPKPERPPSPEVIVDSEEEREDVALQMVGFSNPPVLIKHGKGGKRTVRRLNEDDPVARGMRTQEEEEEPSEAESEITVMNPLSVPIVSAWEKGMEAARALMDKYHVDNDLKANFKLLPDQVEALAAVCKTWLNEEHRGLQLTFTSKKTFVTMMGRFLQAYLQSFAEVTYKHHEPTGCALWLHRCAEIEGELKCLHGSIMINKEHVIEMDVTSENGQRALKEQSSKAKIVKNRWGRNVVQISNTDARCCVHDAACPANQFSGKSCGMFFSEGAKAQVAFKQIKAFMQALYPNAQTGHGHLLMPLRCECNSKPGHAPFLGRQLPKLTPFALSNAEDLDADLISDKSVLASVHHPALIVFQCCNPVYRNSRAQGGGPNCDFKISAPDLLNALVMVRSLWSENFTELPRMVVPEFKWSTKHQYRNVSLPVAHSDARQNPFDF"
|
| 460 |
+
train_df = train_df.loc[train_df['Sequence']!=conflict_seq].reset_index(drop=True)
|
| 461 |
+
log_update(f"\tDropping rows with label mismatch or known error (total={len(train_df[train_df['No Label Conflicts']==False])+1})")
|
| 462 |
+
train_df = train_df.loc[train_df['No Label Conflicts']].reset_index(drop=True)
|
| 463 |
+
|
| 464 |
+
# Make a new label column
|
| 465 |
+
train_df = train_df.drop(columns=["IDP-CRF Label","flDPnn Label"])
|
| 466 |
+
log_update(f"\t\tNew dataset size: {len(train_df)}")
|
| 467 |
+
|
| 468 |
+
######## Final checks
|
| 469 |
+
# Check for any invalid sequences or invalid characters
|
| 470 |
+
cols_of_interest = ['Sequence','Split','Label','IDP-CRF ID','flDPnn ID']
|
| 471 |
+
listlike_dict = check_columns_for_listlike(train_df, cols_of_interest, DELIMITERS)
|
| 472 |
+
|
| 473 |
+
# Check for invalid characters
|
| 474 |
+
train_df['invalid_chars'] = train_df['Sequence'].apply(lambda x: find_invalid_chars(x, VALID_AAS))
|
| 475 |
+
train_df[train_df['invalid_chars'].str.len()>0].sort_values(by='Sequence')
|
| 476 |
+
all_invalid_chars = set().union(*train_df['invalid_chars'])
|
| 477 |
+
log_update(f"\tchecking for invalid characters...\n\t\tset of all invalid characters discovered within train_df: {all_invalid_chars}")
|
| 478 |
+
|
| 479 |
+
# Dropping rows where invalid characters(should be none)
|
| 480 |
+
log_update(f"\tDropping rows with invalid characters (total={len(train_df[train_df['invalid_chars'].str.len()>0])})")
|
| 481 |
+
train_df = train_df.loc[train_df['invalid_chars'].str.len()==0].reset_index(drop=True)
|
| 482 |
+
train_df = train_df.drop(columns=['invalid_chars'])
|
| 483 |
+
log_update(f"\t\tNew dataset size: {len(train_df)}")
|
| 484 |
+
|
| 485 |
+
source_str = train_df['Source'].value_counts().reset_index().rename(columns={'index': 'Source','Source': 'count'}).to_string(index=False)
|
| 486 |
+
source_str = "\t\t" + source_str.replace("\n","\n\t\t")
|
| 487 |
+
log_update(f"\tSources:\n{source_str}")
|
| 488 |
+
return train_df
|
| 489 |
+
|
| 490 |
+
def make_train_and_test_df(train_df, test_df):
|
| 491 |
+
"""
|
| 492 |
+
Combine the training and testing dataframe into one
|
| 493 |
+
"""
|
| 494 |
+
log_update("\nMaking final dataframe with train and test splits")
|
| 495 |
+
# Concatenate proposed train and test
|
| 496 |
+
test_df["Source"] = ["CAID-2_Disorder_NOX"]*len(test_df)
|
| 497 |
+
splits_df = pd.concat([train_df.drop(columns=['No Label Conflicts']),
|
| 498 |
+
test_df.rename(columns={'ID':'CAID-2_Disorder_NOX ID', 'Label': 'CAID-2_Disorder_NOX Label'})])
|
| 499 |
+
split_str = splits_df['Split'].value_counts().reset_index().rename(columns={'index': 'Split','Split': 'count'}).to_string(index=False)
|
| 500 |
+
split_str = "\t\t" + split_str.replace("\n","\n\t\t")
|
| 501 |
+
log_update(f"\tTrain dataset size: {len(train_df)}\n\tTest dataset size: {len(test_df)}\n\tinitial combined dataset size: {len(splits_df)}")
|
| 502 |
+
|
| 503 |
+
# Check for duplicates - if we find any, REMOVE them from train and keep them in test
|
| 504 |
+
duplicates = splits_df[splits_df.duplicated('Sequence')]['Sequence'].unique().tolist()
|
| 505 |
+
n_rows_with_duplicates = len(splits_df[splits_df['Sequence'].isin(duplicates)])
|
| 506 |
+
log_update(f"\t\t{len(duplicates)} duplicated sequences, corresponding to {n_rows_with_duplicates} rows")
|
| 507 |
+
for i, dup in enumerate(duplicates):
|
| 508 |
+
fldpnn_id = splits_df.loc[(splits_df['Sequence']==dup)&(splits_df['Split']=='Train')]['flDPnn ID'].item()
|
| 509 |
+
idp_crf_id = splits_df.loc[(splits_df['Sequence']==dup)&(splits_df['Split']=='Train')]['IDP-CRF ID'].item()
|
| 510 |
+
caid2_disorder_nox_id = splits_df.loc[(splits_df['Sequence']==dup)&(splits_df['Split']=='Test')]['CAID-2_Disorder_NOX ID'].item()
|
| 511 |
+
log_update(f"\t\t\t{i+1}: flDPnn ID: {fldpnn_id}\tIDP-CRF ID: {idp_crf_id}\tCAID-2_Disorder_NOX ID: {caid2_disorder_nox_id}\n\t\t\t\tSequence: {dup}")
|
| 512 |
+
# remove from train and keep in test
|
| 513 |
+
splits_df = splits_df.loc[
|
| 514 |
+
(~splits_df['Sequence'].isin(duplicates)) | # Either the sequence is NOT duplicated, or
|
| 515 |
+
((splits_df['Sequence'].isin(duplicates)) & (splits_df['Split']=='Test')) # Sequence is duplicated, and it's in test set
|
| 516 |
+
].reset_index(drop=True)
|
| 517 |
+
split_str = splits_df['Split'].value_counts().reset_index().rename(columns={'index': 'Split','Split': 'count'}).to_string(index=False)
|
| 518 |
+
split_str = "\t\t" + split_str.replace("\n","\n\t\t")
|
| 519 |
+
log_update(f"\tRemoved duplicate sequences from training split, kept in test split\n\t\tNew dataset size: {len(splits_df)}\n\n{split_str}")
|
| 520 |
+
|
| 521 |
+
# Everything in the train set should have a label; nothing in the test set should
|
| 522 |
+
assert splits_df[splits_df["Label"].isna()]["Split"].value_counts().reset_index()['index'].tolist()==['Test']
|
| 523 |
+
splits_df.loc[
|
| 524 |
+
splits_df["Split"]=="Test","Label"
|
| 525 |
+
] = splits_df.loc[
|
| 526 |
+
splits_df["Split"]=="Test","CAID-2_Disorder_NOX Label"
|
| 527 |
+
]
|
| 528 |
+
splits_df = splits_df.drop(columns=["CAID-2_Disorder_NOX Label"])
|
| 529 |
+
# Make sure there are no na's in label
|
| 530 |
+
assert len(splits_df[splits_df["Label"].isna()])==0
|
| 531 |
+
|
| 532 |
+
# Print out distribution of sources
|
| 533 |
+
source_str = splits_df['Source'].value_counts().reset_index().rename(columns={'index': 'Source','Source': 'count'}).to_string(index=False)
|
| 534 |
+
source_str = "\t\t" + source_str.replace("\n","\n\t\t")
|
| 535 |
+
total_sources = sum(splits_df['Source'].value_counts().reset_index()['Source'])
|
| 536 |
+
assert total_sources == len(splits_df)
|
| 537 |
+
log_update(f"\n\tSource distribution:\n{source_str}\n\n\t\t\t\t\t\tSum: {total_sources}")
|
| 538 |
+
|
| 539 |
+
# Print largest and smallest seq len in each set
|
| 540 |
+
longest_train = max(splits_df[splits_df['Split']=='Train']['Sequence'].apply(lambda x: len(x)).tolist())
|
| 541 |
+
shortest_train = min(splits_df[splits_df['Split']=='Train']['Sequence'].apply(lambda x: len(x)).tolist())
|
| 542 |
+
longest_test = max(splits_df[splits_df['Split']=='Test']['Sequence'].apply(lambda x: len(x)).tolist())
|
| 543 |
+
shortest_test = min(splits_df[splits_df['Split']=='Test']['Sequence'].apply(lambda x: len(x)).tolist())
|
| 544 |
+
log_update(f"\n\tLength distributions...\n\t\tTrain: max={longest_train}\tmin={shortest_train}\n\t\tTest: max={longest_test}\tmin={shortest_test}")
|
| 545 |
+
|
| 546 |
+
# Consolidate the IDs a bit
|
| 547 |
+
splits_df["IDs"] = splits_df.apply(lambda row: get_unique_ids(row),axis=1)
|
| 548 |
+
assert len(splits_df[splits_df["IDs"].isna()])==0
|
| 549 |
+
n_different_ids = len(splits_df.loc[splits_df["IDs"].str.contains(",")])
|
| 550 |
+
log_update(f"\n\tProvided comma-separated IDs in same listed order as Source\n\t\t- train: IDP-CRF first, flDPnn second ({n_different_ids} seqs have multiple distinct IDs)\n\t\t- test: CAID-2_Disorder_NOX")
|
| 551 |
+
|
| 552 |
+
# Keep only desired columns
|
| 553 |
+
splits_df = splits_df[[
|
| 554 |
+
'Sequence','IDs','Split','Source','Label'
|
| 555 |
+
]]
|
| 556 |
+
|
| 557 |
+
return splits_df
|
| 558 |
+
|
| 559 |
+
def main():
|
| 560 |
+
with open_logfile("data_cleaning_log.txt"):
|
| 561 |
+
rawdata_train_test_path = "raw_data/caid2_train_and_test_data"
|
| 562 |
+
# make directory to save processed data
|
| 563 |
+
processeddata_path = "processed_data"
|
| 564 |
+
splits_path = "splits"
|
| 565 |
+
os.makedirs(processeddata_path,exist_ok=True)
|
| 566 |
+
os.makedirs(splits_path,exist_ok=True)
|
| 567 |
+
|
| 568 |
+
# Process CAID-2_Disorder_NOX_Testing_Sequences dataset from fasta file
|
| 569 |
+
caid_path = f"{rawdata_train_test_path}/CAID-2_Disorder_NOX_Testing_Sequences.fasta"
|
| 570 |
+
caid_df = process_caid2_disorder_nox_test(caid_path)
|
| 571 |
+
caid_df.to_csv(f"{processeddata_path}/CAID-2_Disorder_NOX_Processed.csv", index=False)
|
| 572 |
+
|
| 573 |
+
# Process fldpnn Training and Validation Datasets
|
| 574 |
+
fldpnn_train_path = f"{rawdata_train_test_path}/flDPnn_Training_Dataset.txt"
|
| 575 |
+
fldpnn_val_path = f"{rawdata_train_test_path}/flDPnn_Validation_Annotation.txt"
|
| 576 |
+
fldpnn_train_df = process_fldpnn(fldpnn_train_path, split="training")
|
| 577 |
+
fldpnn_val_df = process_fldpnn(fldpnn_val_path, split="validation")
|
| 578 |
+
fldpnn_train_df.to_csv(f"{processeddata_path}/flDPnn_Training_Dataset.csv", index=False)
|
| 579 |
+
fldpnn_val_df.to_csv(f"{processeddata_path}/flDPnn_Validation_Dataset.csv", index=False)
|
| 580 |
+
# Combine train and val
|
| 581 |
+
fldpnn_df = combine_fldpnn_train_val(fldpnn_train_df, fldpnn_val_df)
|
| 582 |
+
|
| 583 |
+
# Process IDP-CRF_Training_Dataset
|
| 584 |
+
idp_crf_train_path = f"{rawdata_train_test_path}/IDP-CRF_Training_Dataset.txt"
|
| 585 |
+
idp_crf_df= process_idp_crf_train(idp_crf_train_path)
|
| 586 |
+
idp_crf_df.to_csv(f"{processeddata_path}/IDP-CRF_Training_Dataset.csv", index=False)
|
| 587 |
+
|
| 588 |
+
# Merge
|
| 589 |
+
train_df = make_train_df(fldpnn_df, idp_crf_df)
|
| 590 |
+
|
| 591 |
+
# Make a full splits file
|
| 592 |
+
splits_df = make_train_and_test_df(train_df, caid_df)
|
| 593 |
+
final_train_df = splits_df.loc[splits_df['Split']=='Train'].reset_index(drop=True)
|
| 594 |
+
final_test_df = splits_df.loc[splits_df['Split']=='Test'].reset_index(drop=True)
|
| 595 |
+
|
| 596 |
+
# Save final files
|
| 597 |
+
final_train_df.to_csv(f"{splits_path}/train_df.csv", index=False)
|
| 598 |
+
final_test_df.to_csv(f"{splits_path}/test_df.csv", index=False)
|
| 599 |
+
|
| 600 |
+
# Process the caid competition results and save them in a more accessible format
|
| 601 |
+
processed_caid2_df = pd.read_csv(f"{processeddata_path}/CAID-2_Disorder_NOX_Processed.csv")
|
| 602 |
+
parse_all_caid2_results(processed_caid2_df)
|
| 603 |
+
|
| 604 |
+
# Process data for visualizing fusion oncoproteins
|
| 605 |
+
# Scrape FusionPDB
|
| 606 |
+
scrape_fusionpdb_level_2_3()
|
| 607 |
+
# Process the structures that we downloaded from scraping
|
| 608 |
+
process_fusions_and_hts()
|
| 609 |
+
|
| 610 |
+
# Now, figure out which structures are in the test set and isolate those for benchmarking in splits/fusion_bench_df.csv
|
| 611 |
+
fusion_test_set = pd.read_csv("../../data/splits/test_df.csv")
|
| 612 |
+
# columns are sequence, member length, snp_probabilities
|
| 613 |
+
fusion_test_set = set(fusion_test_set['sequence'].tolist())
|
| 614 |
+
log_update(f"\nFinding level 2 and 3 fusion structures that are in the FusOn-pLM test set...\n\tTest set size: {len(fusion_test_set)} seqs")
|
| 615 |
+
level_2_3_info = pd.read_csv('processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv')
|
| 616 |
+
# there are duplicate sequences in here so drop duplicates randomly
|
| 617 |
+
level_2_3_seqs = level_2_3_info.drop_duplicates('Fusion_Seq').reset_index(drop=True)
|
| 618 |
+
level_2_3_seqs = set(level_2_3_seqs.loc[
|
| 619 |
+
level_2_3_info['Fusion_pLDDT'].notna() # make sure we've got a structure
|
| 620 |
+
]['Fusion_Seq'].tolist())
|
| 621 |
+
# if it has a structure, it's in the test set, and it's not in the caid train set, we can benchmark with it
|
| 622 |
+
test_benchmark_seqs = fusion_test_set.intersection(level_2_3_seqs)
|
| 623 |
+
log_update(f"\tTotal fusion proteins in the FusOn-pLM test set: {len(test_benchmark_seqs)}")
|
| 624 |
+
caid_train_set = set(pd.read_csv('splits/train_df.csv')['Sequence'].tolist())
|
| 625 |
+
test_benchmark_seqs = test_benchmark_seqs.difference(caid_train_set) # subtract off the caid train set to be sure
|
| 626 |
+
log_update(f"\tTotal fusion proteins in the FusOn-pLM test set and NOT in the CAID train set: {len(test_benchmark_seqs)}")
|
| 627 |
+
|
| 628 |
+
# Finally, make a dataframe structured like train_df and test_df. Columns are: Sequence,IDs,Split,Source,Label
|
| 629 |
+
# Let's make the IDs FusionGID
|
| 630 |
+
test_benchmark_df = pd.DataFrame(
|
| 631 |
+
data = {'Sequence': list(test_benchmark_seqs)}
|
| 632 |
+
|
| 633 |
+
)
|
| 634 |
+
seq_id_dict = dict(zip(level_2_3_info['Fusion_Seq'],level_2_3_info['FusionGID']))
|
| 635 |
+
seq_plddts_dict = dict(zip(level_2_3_info['Fusion_Seq'],level_2_3_info['Fusion_AA_pLDDTs']))
|
| 636 |
+
test_benchmark_df['IDs'] = test_benchmark_df['Sequence'].map(seq_id_dict)
|
| 637 |
+
test_benchmark_df['Split'] = ['Fusion_Benchmark']*len(test_benchmark_df)
|
| 638 |
+
test_benchmark_df['Source'] = ['FusionPDB_AlphaFold2']*len(test_benchmark_df)
|
| 639 |
+
test_benchmark_df['Label'] = test_benchmark_df['Sequence'].map(seq_plddts_dict)
|
| 640 |
+
# convert label to 1 or 0
|
| 641 |
+
test_benchmark_df['Label'] = test_benchmark_df['Label'].apply(lambda x: x.split(","))
|
| 642 |
+
test_benchmark_df['Label'] = test_benchmark_df['Label'].apply(lambda x: [float(y) for y in x]) # make it a float list of pLDDTs
|
| 643 |
+
test_benchmark_df['Label'] = test_benchmark_df['Label'].apply(lambda x: ['1' if y < 68.8 else '0' for y in x]) # disordered if pLDDT is < 68.8, accoridng to AlphaFold-pLDDT published threshold
|
| 644 |
+
test_benchmark_df['Label'] = test_benchmark_df['Label'].apply(lambda x: ''.join(x)) # change ['1','1','0''] to '110'
|
| 645 |
+
|
| 646 |
+
# check lengths
|
| 647 |
+
test_benchmark_df['SeqLen'] = test_benchmark_df['Sequence'].apply(lambda x: len(x))
|
| 648 |
+
test_benchmark_df['LabelLen'] = test_benchmark_df['Label'].apply(lambda x: len(x))
|
| 649 |
+
log_update(f"\tAll seq lengths and label lengths match: {(test_benchmark_df['SeqLen']==test_benchmark_df['LabelLen']).all()}")
|
| 650 |
+
test_benchmark_df = test_benchmark_df.drop(columns=['SeqLen','LabelLen'])
|
| 651 |
+
|
| 652 |
+
# convert to string
|
| 653 |
+
test_benchmark_df_str = test_benchmark_df.head(10)
|
| 654 |
+
test_benchmark_df_str['Sequence'] = test_benchmark_df_str['Sequence'].apply(lambda x: x[0:10]+'...')
|
| 655 |
+
test_benchmark_df_str['Label'] = test_benchmark_df_str['Label'].apply(lambda x: x[0:10]+'...')
|
| 656 |
+
test_benchmark_df_str = test_benchmark_df_str.to_string(index=False)
|
| 657 |
+
test_benchmark_df_str = "\t" + test_benchmark_df_str.replace("\n","\n\t")
|
| 658 |
+
log_update(f"\nPreview of benchmarking set:\n{test_benchmark_df_str}")
|
| 659 |
+
test_benchmark_df.to_csv('splits/fusion_bench_df.csv',index=False)
|
| 660 |
+
|
| 661 |
+
# Add the benchmarking sequences to split
|
| 662 |
+
log_update(f"\nAdding benchmarking sequences to splits_df.csv:\n\tLength before adding bench seqs: {len(splits_df)}")
|
| 663 |
+
splits_df = pd.concat([splits_df,test_benchmark_df])
|
| 664 |
+
log_update(f"\tLength after adding bench seqs: {len(splits_df)}")
|
| 665 |
+
split_str = splits_df['Split'].value_counts().reset_index().rename(columns={'index': 'Split','Split': 'count'}).to_string(index=False)
|
| 666 |
+
split_str = "\t" + split_str.replace("\n","\n\t")
|
| 667 |
+
log_update(f"Distribution among splits:\n{split_str}")
|
| 668 |
+
splits_df.to_csv(f"{splits_path}/splits.csv",index=False)
|
| 669 |
+
|
| 670 |
+
if __name__ == "__main__":
|
| 671 |
+
main()
|
fuson_plm/benchmarking/caid/color_disordered_residues.ipynb
ADDED
|
@@ -0,0 +1,849 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
{
|
| 2 |
+
"cells": [
|
| 3 |
+
{
|
| 4 |
+
"cell_type": "markdown",
|
| 5 |
+
"metadata": {
|
| 6 |
+
"id": "FJd6a9gdZNjG"
|
| 7 |
+
},
|
| 8 |
+
"source": [
|
| 9 |
+
"### Imports"
|
| 10 |
+
]
|
| 11 |
+
},
|
| 12 |
+
{
|
| 13 |
+
"cell_type": "code",
|
| 14 |
+
"execution_count": 1,
|
| 15 |
+
"metadata": {},
|
| 16 |
+
"outputs": [],
|
| 17 |
+
"source": [
|
| 18 |
+
"## Put path to model predictions you'd like to use for benchmarking\n",
|
| 19 |
+
"path_to_model_predictions = \"trained_models/fuson_plm/best/caid_hyperparam_screen_fusion_benchmark_probs.csv\""
|
| 20 |
+
]
|
| 21 |
+
},
|
| 22 |
+
{
|
| 23 |
+
"cell_type": "code",
|
| 24 |
+
"execution_count": null,
|
| 25 |
+
"metadata": {},
|
| 26 |
+
"outputs": [],
|
| 27 |
+
"source": [
|
| 28 |
+
"!pip install torch pandas numpy py3Dmol scikit-learn"
|
| 29 |
+
]
|
| 30 |
+
},
|
| 31 |
+
{
|
| 32 |
+
"cell_type": "code",
|
| 33 |
+
"execution_count": 3,
|
| 34 |
+
"metadata": {
|
| 35 |
+
"id": "ZEWZVc9lUxjI"
|
| 36 |
+
},
|
| 37 |
+
"outputs": [],
|
| 38 |
+
"source": [
|
| 39 |
+
"import torch\n",
|
| 40 |
+
"import torch.nn as nn\n",
|
| 41 |
+
"\n",
|
| 42 |
+
"import pickle\n",
|
| 43 |
+
"import pandas as pd\n",
|
| 44 |
+
"import numpy as np\n",
|
| 45 |
+
"\n",
|
| 46 |
+
"import py3Dmol\n",
|
| 47 |
+
"\n",
|
| 48 |
+
"from sklearn.metrics import accuracy_score, precision_score, recall_score, f1_score, roc_auc_score, precision_recall_curve, average_precision_score"
|
| 49 |
+
]
|
| 50 |
+
},
|
| 51 |
+
{
|
| 52 |
+
"cell_type": "code",
|
| 53 |
+
"execution_count": 4,
|
| 54 |
+
"metadata": {
|
| 55 |
+
"id": "Uu0z_JYYUzMC"
|
| 56 |
+
},
|
| 57 |
+
"outputs": [],
|
| 58 |
+
"source": [
|
| 59 |
+
"# Define paths and dataframes that we will need \n",
|
| 60 |
+
"fusion_benchmark_set = pd.read_csv('splits/fusion_bench_df.csv')\n",
|
| 61 |
+
"model_predictions = pd.read_csv(path_to_model_predictions)\n",
|
| 62 |
+
"fusion_structure_data = pd.read_csv('processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv')\n",
|
| 63 |
+
"fusion_structure_data['Fusion_Structure_Link'] = fusion_structure_data['Fusion_Structure_Link'].apply(lambda x: x.split('/')[-1])\n",
|
| 64 |
+
"fusion_structure_folder = \"raw_data/fusionpdb/structures\""
|
| 65 |
+
]
|
| 66 |
+
},
|
| 67 |
+
{
|
| 68 |
+
"cell_type": "code",
|
| 69 |
+
"execution_count": null,
|
| 70 |
+
"metadata": {},
|
| 71 |
+
"outputs": [],
|
| 72 |
+
"source": [
|
| 73 |
+
"# merge fusion data with seq ids \n",
|
| 74 |
+
"fuson_db = pd.read_csv('../../data/fuson_db.csv')\n",
|
| 75 |
+
"print(fuson_db.columns)\n",
|
| 76 |
+
"fuson_db = fuson_db[['aa_seq','seq_id']].rename(columns={'aa_seq':'Fusion_Seq'})\n",
|
| 77 |
+
"print(f\"Length of fusion structure data before merge on seqid: {len(fusion_structure_data)}\")\n",
|
| 78 |
+
"fusion_structure_data = pd.merge(\n",
|
| 79 |
+
" fusion_structure_data,\n",
|
| 80 |
+
" fuson_db,\n",
|
| 81 |
+
" on='Fusion_Seq',\n",
|
| 82 |
+
" how='inner'\n",
|
| 83 |
+
")\n",
|
| 84 |
+
"print(f\"Length of fusion structure data after merge on seqid: {len(fusion_structure_data)}\")\n",
|
| 85 |
+
"fusion_structure_data"
|
| 86 |
+
]
|
| 87 |
+
},
|
| 88 |
+
{
|
| 89 |
+
"cell_type": "code",
|
| 90 |
+
"execution_count": null,
|
| 91 |
+
"metadata": {},
|
| 92 |
+
"outputs": [],
|
| 93 |
+
"source": [
|
| 94 |
+
"# merge fusion structure data with top swissprot alignments\n",
|
| 95 |
+
"swissprot_top_alignments = pd.read_csv(\"../../data/blast/blast_outputs/swissprot_top_alignments.csv\")\n",
|
| 96 |
+
"fusion_structure_data = pd.merge(\n",
|
| 97 |
+
" fusion_structure_data,\n",
|
| 98 |
+
" swissprot_top_alignments,\n",
|
| 99 |
+
" on=\"seq_id\",\n",
|
| 100 |
+
" how=\"left\"\n",
|
| 101 |
+
")\n",
|
| 102 |
+
"fusion_structure_data.head()"
|
| 103 |
+
]
|
| 104 |
+
},
|
| 105 |
+
{
|
| 106 |
+
"cell_type": "code",
|
| 107 |
+
"execution_count": null,
|
| 108 |
+
"metadata": {},
|
| 109 |
+
"outputs": [],
|
| 110 |
+
"source": [
|
| 111 |
+
"print(list(fusion_structure_data.columns))"
|
| 112 |
+
]
|
| 113 |
+
},
|
| 114 |
+
{
|
| 115 |
+
"cell_type": "markdown",
|
| 116 |
+
"metadata": {
|
| 117 |
+
"id": "zORkLJztZWp9"
|
| 118 |
+
},
|
| 119 |
+
"source": [
|
| 120 |
+
"### Prepare data"
|
| 121 |
+
]
|
| 122 |
+
},
|
| 123 |
+
{
|
| 124 |
+
"cell_type": "code",
|
| 125 |
+
"execution_count": 8,
|
| 126 |
+
"metadata": {
|
| 127 |
+
"id": "JKbEOWP390ba"
|
| 128 |
+
},
|
| 129 |
+
"outputs": [],
|
| 130 |
+
"source": [
|
| 131 |
+
"def interpolate_color(value, start_color, end_color):\n",
|
| 132 |
+
" return [(1 - value) * start + value * end for start, end in zip(start_color, end_color)]\n",
|
| 133 |
+
"\n",
|
| 134 |
+
"# Define the color points for the gradient\n",
|
| 135 |
+
"\n",
|
| 136 |
+
"def get_color(value):\n",
|
| 137 |
+
" colors = [\n",
|
| 138 |
+
" (0, 0, 0.545), # Dark Blue\n",
|
| 139 |
+
" (0.678, 0.847, 0.902), # Light Blue\n",
|
| 140 |
+
" (1, 1, 0), # Yellow\n",
|
| 141 |
+
" (1, 0.65, 0) # Orange\n",
|
| 142 |
+
" ]\n",
|
| 143 |
+
"\n",
|
| 144 |
+
" if value <= 10:\n",
|
| 145 |
+
" return interpolate_color(value / 10, colors[0], colors[1])\n",
|
| 146 |
+
" elif value <= 30:\n",
|
| 147 |
+
" return interpolate_color((value - 10) / 20, colors[1], colors[2])\n",
|
| 148 |
+
" elif value <= 50:\n",
|
| 149 |
+
" return interpolate_color((value - 30) / 20, colors[2], colors[3])\n",
|
| 150 |
+
" else:\n",
|
| 151 |
+
" return interpolate_color((value - 50) / 50, colors[3], colors[3])\n",
|
| 152 |
+
"\n",
|
| 153 |
+
"def color_by_disorder(cif_file, disorder_values):\n",
|
| 154 |
+
" # Create viewer\n",
|
| 155 |
+
" viewer = py3Dmol.view()\n",
|
| 156 |
+
"\n",
|
| 157 |
+
" # Load CIF file\n",
|
| 158 |
+
" with open(cif_file, 'r') as f:\n",
|
| 159 |
+
" cif_data = f.read()\n",
|
| 160 |
+
"\n",
|
| 161 |
+
" # Add structure\n",
|
| 162 |
+
" viewer.addModel(cif_data, 'cif')\n",
|
| 163 |
+
"\n",
|
| 164 |
+
" # Normalize disorder values to be between 0 and 100\n",
|
| 165 |
+
" min_disorder = min(disorder_values)\n",
|
| 166 |
+
" max_disorder = max(disorder_values)\n",
|
| 167 |
+
" normalized_disorder = [(val - min_disorder) / (max_disorder - min_disorder) * 100 for val in disorder_values]\n",
|
| 168 |
+
"\n",
|
| 169 |
+
" # Apply colors based on normalized disorder values\n",
|
| 170 |
+
" for i, value in enumerate(normalized_disorder, start=1):\n",
|
| 171 |
+
" mix_color = get_color(value)\n",
|
| 172 |
+
" rgb_color = f'rgb({int(mix_color[0]*255)}, {int(mix_color[1]*255)}, {int(mix_color[2]*255)})'\n",
|
| 173 |
+
" viewer.setStyle({'resi': i}, {'cartoon': {'color': rgb_color}})\n",
|
| 174 |
+
"\n",
|
| 175 |
+
" # Show viewer\n",
|
| 176 |
+
" viewer.zoomTo()\n",
|
| 177 |
+
" return viewer.show()"
|
| 178 |
+
]
|
| 179 |
+
},
|
| 180 |
+
{
|
| 181 |
+
"cell_type": "code",
|
| 182 |
+
"execution_count": 9,
|
| 183 |
+
"metadata": {},
|
| 184 |
+
"outputs": [],
|
| 185 |
+
"source": [
|
| 186 |
+
"# AlphaFold2 Database colors\n",
|
| 187 |
+
"def get_alphafold2_color(pLDDT):\n",
|
| 188 |
+
" if pLDDT > 90: # darkblue\n",
|
| 189 |
+
" return (13, 87, 211)\n",
|
| 190 |
+
" elif pLDDT > 70: # lightblue\n",
|
| 191 |
+
" return (106, 203, 241)\n",
|
| 192 |
+
" elif pLDDT > 50: # yellow\n",
|
| 193 |
+
" return (254, 217, 54)\n",
|
| 194 |
+
" else: # orange\n",
|
| 195 |
+
" return (253, 125, 77)\n",
|
| 196 |
+
"\n",
|
| 197 |
+
"def color_by_pLDDT(cif_file, plddt_values):\n",
|
| 198 |
+
" viewer = py3Dmol.view(width=400, height=400)\n",
|
| 199 |
+
" viewer.addModel(open(cif_file, 'r').read(), 'cif')\n",
|
| 200 |
+
"\n",
|
| 201 |
+
" # Apply colors based on normalized disorder values\n",
|
| 202 |
+
" for i, value in enumerate(plddt_values, start=1):\n",
|
| 203 |
+
" mix_color = get_alphafold2_color(value)\n",
|
| 204 |
+
" rgb_color = f'rgb({int(mix_color[0])}, {int(mix_color[1])}, {int(mix_color[2])})'\n",
|
| 205 |
+
" viewer.setStyle({'resi': i}, {'cartoon': {'color': rgb_color}})\n",
|
| 206 |
+
"\n",
|
| 207 |
+
" viewer.zoomTo()\n",
|
| 208 |
+
" viewer.show()\n",
|
| 209 |
+
" \n",
|
| 210 |
+
"def color_by_head_tail(cif_file, head_coords, tail_coords):\n",
|
| 211 |
+
" \"\"\"\n",
|
| 212 |
+
" head_coords and tail_coords are list of [start_psn, end_psn] inclusive. 1-indexed.\n",
|
| 213 |
+
" \"\"\"\n",
|
| 214 |
+
" head_resi_nos = list(range(head_coords[0], head_coords[1]+1))\n",
|
| 215 |
+
" tail_resi_nos = list(range(tail_coords[0], tail_coords[1]+1))\n",
|
| 216 |
+
" \n",
|
| 217 |
+
" viewer = py3Dmol.view(width=400, height=400)\n",
|
| 218 |
+
" viewer.addModel(open(cif_file, 'r').read(), 'cif')\n",
|
| 219 |
+
"\n",
|
| 220 |
+
" viewer.setStyle({'cartoon': {'color': '#cf9dfa'}}) # fusion color purple\n",
|
| 221 |
+
" viewer.setStyle({'resi': head_resi_nos}, {'cartoon': {'color': '#eb8888'}})\n",
|
| 222 |
+
" viewer.setStyle({'resi': tail_resi_nos}, {'cartoon': {'color': '#5fa3e3'}})\n",
|
| 223 |
+
"\n",
|
| 224 |
+
" viewer.zoomTo()\n",
|
| 225 |
+
" viewer.show()\n",
|
| 226 |
+
"\n",
|
| 227 |
+
"def two_coloring_viewer_grid(cif_file, plddt_values, head_coords, tail_coords):\n",
|
| 228 |
+
" head_resi_nos = list(range(head_coords[0], head_coords[1]+1))\n",
|
| 229 |
+
" tail_resi_nos = list(range(tail_coords[0], tail_coords[1]+1))\n",
|
| 230 |
+
" \n",
|
| 231 |
+
" view = py3Dmol.view(width=800, height=400, viewergrid=(1,2))\n",
|
| 232 |
+
" view.addModel(open(cif_file, 'r').read(), 'cif')\n",
|
| 233 |
+
"\n",
|
| 234 |
+
" # Apply colors based on normalized disorder values\n",
|
| 235 |
+
" for i, value in enumerate(plddt_values, start=1):\n",
|
| 236 |
+
" mix_color = get_alphafold2_color(value)\n",
|
| 237 |
+
" rgb_color = f'rgb({int(mix_color[0])}, {int(mix_color[1])}, {int(mix_color[2])})'\n",
|
| 238 |
+
" view.setStyle({'resi': i}, {'cartoon': {'color': rgb_color}},viewer=(0,0))\n",
|
| 239 |
+
"\n",
|
| 240 |
+
" view.setStyle({'cartoon': {'color': '#cf9dfa'}},viewer=(0,1)) # fusion color purple\n",
|
| 241 |
+
" view.setStyle({'resi': head_resi_nos}, {'cartoon': {'color': '#eb8888'}},viewer=(0,1))\n",
|
| 242 |
+
" view.setStyle({'resi': tail_resi_nos}, {'cartoon': {'color': '#5fa3e3'}},viewer=(0,1))\n",
|
| 243 |
+
"\n",
|
| 244 |
+
" view.zoomTo()\n",
|
| 245 |
+
" view.render()\n",
|
| 246 |
+
" view.show()\n",
|
| 247 |
+
"\n",
|
| 248 |
+
"import os\n",
|
| 249 |
+
"def four_coloring_viewer_grid(cif_file, plddt_values, disorder_values, head_coords, tail_coords, seq_id=None, save_normalized_propensities=False):\n",
|
| 250 |
+
" head_resi_nos = list(range(head_coords[0], head_coords[1]+1))\n",
|
| 251 |
+
" tail_resi_nos = list(range(tail_coords[0], tail_coords[1]+1))\n",
|
| 252 |
+
" \n",
|
| 253 |
+
" view = py3Dmol.view(width=800, height=800, viewergrid=(2,2))\n",
|
| 254 |
+
" view.addModel(open(cif_file, 'r').read(), 'cif')\n",
|
| 255 |
+
"\n",
|
| 256 |
+
" # (0,0): apply AF2 colors based on normalized disorder values\n",
|
| 257 |
+
" for i, value in enumerate(plddt_values, start=1):\n",
|
| 258 |
+
" mix_color = get_alphafold2_color(value)\n",
|
| 259 |
+
" rgb_color = f'rgb({int(mix_color[0])}, {int(mix_color[1])}, {int(mix_color[2])})'\n",
|
| 260 |
+
" view.setStyle({'resi': i}, {'cartoon': {'color': rgb_color}},viewer=(0,0))\n",
|
| 261 |
+
"\n",
|
| 262 |
+
" # (0,1): apply our colors based on normalized disorder values\n",
|
| 263 |
+
" min_plddt = min(plddt_values)\n",
|
| 264 |
+
" max_plddt = max(plddt_values)\n",
|
| 265 |
+
" normalized_inverse_plddts = [(val - min_plddt) / (max_plddt - min_plddt) * 100 for val in plddt_values]\n",
|
| 266 |
+
" normalized_inverse_plddts = [100 - val for val in normalized_inverse_plddts]\n",
|
| 267 |
+
"\n",
|
| 268 |
+
" for i, value in enumerate(normalized_inverse_plddts, start=1):\n",
|
| 269 |
+
" mix_color = get_color(value)\n",
|
| 270 |
+
" rgb_color = f'rgb({int(mix_color[0]*255)}, {int(mix_color[1]*255)}, {int(mix_color[2]*255)})'\n",
|
| 271 |
+
" view.setStyle({'resi': i}, {'cartoon': {'color': rgb_color}},viewer=(0,1))\n",
|
| 272 |
+
"\n",
|
| 273 |
+
" # (1,0): apply colors based on head and tail segments\n",
|
| 274 |
+
" view.setStyle({'cartoon': {'color': '#cf9dfa'}},viewer=(1,0)) # fusion color purple\n",
|
| 275 |
+
" view.setStyle({'resi': head_resi_nos}, {'cartoon': {'color': '#eb8888'}},viewer=(1,0))\n",
|
| 276 |
+
" view.setStyle({'resi': tail_resi_nos}, {'cartoon': {'color': '#5fa3e3'}},viewer=(1,0))\n",
|
| 277 |
+
" \n",
|
| 278 |
+
" ## (1,1): apply colors based on disorder predictions\n",
|
| 279 |
+
" # Normalize disorder values to be between 0 and 100\n",
|
| 280 |
+
" min_disorder = min(disorder_values)\n",
|
| 281 |
+
" max_disorder = max(disorder_values)\n",
|
| 282 |
+
" normalized_disorder = [(val - min_disorder) / (max_disorder - min_disorder) * 100 for val in disorder_values]\n",
|
| 283 |
+
"\n",
|
| 284 |
+
" # if we're saving normalized propensities, then save them here\n",
|
| 285 |
+
" if save_normalized_propensities:\n",
|
| 286 |
+
" normalized_disorder_df = pd.DataFrame(\n",
|
| 287 |
+
" data={\n",
|
| 288 |
+
" 'seq_id': seq_id,\n",
|
| 289 |
+
" 'normalized_disorder': [normalized_disorder]\n",
|
| 290 |
+
" }\n",
|
| 291 |
+
" )\n",
|
| 292 |
+
" normalized_disorder_df['normalized_disorder'] = normalized_disorder_df['normalized_disorder'].apply(lambda x: [round(y,6) for y in x])\n",
|
| 293 |
+
" os.makedirs(\"disorder_coloring_data\",exist_ok=True)\n",
|
| 294 |
+
" fname = \"disorder_coloring_data/normalized_disorder_propensities_source_data.csv\"\n",
|
| 295 |
+
" if os.path.isfile(fname):\n",
|
| 296 |
+
" normalized_disorder_df.to_csv(fname,index=False,mode='a',header=False)\n",
|
| 297 |
+
" else:\n",
|
| 298 |
+
" normalized_disorder_df.to_csv(fname,index=False)\n",
|
| 299 |
+
" # Apply colors based on normalized disorder values\n",
|
| 300 |
+
" for i, value in enumerate(normalized_disorder, start=1):\n",
|
| 301 |
+
" mix_color = get_color(value)\n",
|
| 302 |
+
" rgb_color = f'rgb({int(mix_color[0]*255)}, {int(mix_color[1]*255)}, {int(mix_color[2]*255)})'\n",
|
| 303 |
+
" view.setStyle({'resi': i}, {'cartoon': {'color': rgb_color}},viewer=(1,1))\n",
|
| 304 |
+
"\n",
|
| 305 |
+
" view.zoomTo()\n",
|
| 306 |
+
" view.render()\n",
|
| 307 |
+
" view.show()"
|
| 308 |
+
]
|
| 309 |
+
},
|
| 310 |
+
{
|
| 311 |
+
"cell_type": "markdown",
|
| 312 |
+
"metadata": {
|
| 313 |
+
"id": "SiTAKvW9aQvg"
|
| 314 |
+
},
|
| 315 |
+
"source": [
|
| 316 |
+
"### Save disorder prediction results"
|
| 317 |
+
]
|
| 318 |
+
},
|
| 319 |
+
{
|
| 320 |
+
"cell_type": "code",
|
| 321 |
+
"execution_count": null,
|
| 322 |
+
"metadata": {
|
| 323 |
+
"id": "Y6Pxxyk5U_un"
|
| 324 |
+
},
|
| 325 |
+
"outputs": [],
|
| 326 |
+
"source": [
|
| 327 |
+
"model_predictions"
|
| 328 |
+
]
|
| 329 |
+
},
|
| 330 |
+
{
|
| 331 |
+
"cell_type": "code",
|
| 332 |
+
"execution_count": null,
|
| 333 |
+
"metadata": {},
|
| 334 |
+
"outputs": [],
|
| 335 |
+
"source": [
|
| 336 |
+
"model_predictions_labeled = pd.merge(model_predictions,fusion_benchmark_set.rename(columns={'Sequence':'sequence'}),on='sequence',how='inner')\n",
|
| 337 |
+
"model_predictions_labeled = pd.merge(model_predictions_labeled, \n",
|
| 338 |
+
" fusion_structure_data[['FusionGene','Fusion_Seq','Fusion_Structure_Link','Fusion_pLDDT','Fusion_AA_pLDDTs',\n",
|
| 339 |
+
" 'top_hg_UniProtID', 'top_hg_UniProt_isoform', 'top_hg_UniProt_fus_indices', 'top_tg_UniProtID', 'top_tg_UniProt_isoform', \n",
|
| 340 |
+
" 'top_tg_UniProt_fus_indices', 'top_UniProtID', 'top_UniProt_isoform', 'top_UniProt_fus_indices', 'top_UniProt_nIdentities', \n",
|
| 341 |
+
" 'top_UniProt_nPositives']].rename(\n",
|
| 342 |
+
" columns={'Fusion_Seq': 'sequence'}\n",
|
| 343 |
+
" ),\n",
|
| 344 |
+
" on='sequence',\n",
|
| 345 |
+
" how='left')\n",
|
| 346 |
+
"model_predictions_labeled['length'] = model_predictions_labeled['sequence'].str.len()\n",
|
| 347 |
+
"seq_id_dict = dict(zip(fuson_db['Fusion_Seq'],fuson_db['seq_id']))\n",
|
| 348 |
+
"model_predictions_labeled['seq_id'] = model_predictions_labeled['sequence'].map(seq_id_dict)\n",
|
| 349 |
+
"model_predictions_labeled['Fusion_Structure_Link'] = model_predictions_labeled['Fusion_Structure_Link'].apply(lambda x: x.split('/')[-1])\n",
|
| 350 |
+
"model_predictions_labeled"
|
| 351 |
+
]
|
| 352 |
+
},
|
| 353 |
+
{
|
| 354 |
+
"cell_type": "code",
|
| 355 |
+
"execution_count": 12,
|
| 356 |
+
"metadata": {},
|
| 357 |
+
"outputs": [],
|
| 358 |
+
"source": [
|
| 359 |
+
"# calculate AUROC and AUPRC for each sequence\n",
|
| 360 |
+
"def calc_metrics(row):\n",
|
| 361 |
+
" probs = row['prob_1']\n",
|
| 362 |
+
" probs = [float(y) for y in probs.split(',')]\n",
|
| 363 |
+
" true_labels = row['Label']\n",
|
| 364 |
+
" true_labels = [int(y) for y in list(true_labels)]\n",
|
| 365 |
+
" pred_labels = row['pred_labels']\n",
|
| 366 |
+
" pred_labels = [int(y) for y in list(pred_labels)]\n",
|
| 367 |
+
" \n",
|
| 368 |
+
" # Calculate AUROC\n",
|
| 369 |
+
" # Calculate AUPRC\n",
|
| 370 |
+
" # Calculate all the other stats based on the predicted labels\n",
|
| 371 |
+
" \n",
|
| 372 |
+
" flat_binary_preds = np.array(pred_labels)\n",
|
| 373 |
+
" flat_prob_preds = np.array(probs)\n",
|
| 374 |
+
" flat_labels = np.array(true_labels)\n",
|
| 375 |
+
"\n",
|
| 376 |
+
" accuracy = accuracy_score(flat_labels, flat_binary_preds)\n",
|
| 377 |
+
" precision = precision_score(flat_labels, flat_binary_preds)\n",
|
| 378 |
+
" recall = recall_score(flat_labels, flat_binary_preds)\n",
|
| 379 |
+
" f1 = f1_score(flat_labels, flat_binary_preds)\n",
|
| 380 |
+
" try:\n",
|
| 381 |
+
" roc_auc = roc_auc_score(flat_labels, flat_prob_preds)\n",
|
| 382 |
+
" except:\n",
|
| 383 |
+
" roc_auc = np.nan\n",
|
| 384 |
+
" \n",
|
| 385 |
+
" try:\n",
|
| 386 |
+
" auprc = average_precision_score(flat_labels, flat_prob_preds)\n",
|
| 387 |
+
" except: \n",
|
| 388 |
+
" auprc = np.nan\n",
|
| 389 |
+
" \n",
|
| 390 |
+
" return pd.Series({\n",
|
| 391 |
+
" 'Accuracy': round(accuracy,3),\n",
|
| 392 |
+
" 'Precision': round(precision,3),\n",
|
| 393 |
+
" 'Recall': round(recall,3),\n",
|
| 394 |
+
" 'F1': round(f1,3),\n",
|
| 395 |
+
" 'AUROC': round(roc_auc,3) if not(np.isnan(roc_auc)) else roc_auc,\n",
|
| 396 |
+
" 'AUPRC': round(auprc,3) if not(np.isnan(auprc)) else auprc,\n",
|
| 397 |
+
" })\n",
|
| 398 |
+
" "
|
| 399 |
+
]
|
| 400 |
+
},
|
| 401 |
+
{
|
| 402 |
+
"cell_type": "code",
|
| 403 |
+
"execution_count": null,
|
| 404 |
+
"metadata": {},
|
| 405 |
+
"outputs": [],
|
| 406 |
+
"source": [
|
| 407 |
+
"model_predictions_labeled[['Accuracy','Precision','Recall','F1','AUROC','AUPRC']] = model_predictions_labeled.apply(lambda row: calc_metrics(row),axis=1)\n",
|
| 408 |
+
"model_predictions_labeled = model_predictions_labeled.sort_values(by=['AUROC','F1','AUPRC','Accuracy','Precision','Recall'],ascending=[False,False,False,False,False,False]).reset_index(drop=True)\n",
|
| 409 |
+
"model_predictions_labeled['pcnt_disordered'] = round(100*model_predictions_labeled['Label'].apply(lambda x: sum([int(y) for y in list(x)]))/model_predictions_labeled['sequence'].str.len(),2)\n",
|
| 410 |
+
"model_predictions_labeled['pred_pcnt_disordered'] = round(100*model_predictions_labeled['pred_labels'].apply(lambda x: sum([int(y) for y in list(x)]))/model_predictions_labeled['sequence'].str.len(),2)\n",
|
| 411 |
+
"model_predictions_labeled[['sequence','length','FusionGene','Fusion_pLDDT','pcnt_disordered','pred_pcnt_disordered','AUROC','F1','AUPRC','Accuracy','Precision','Recall']]"
|
| 412 |
+
]
|
| 413 |
+
},
|
| 414 |
+
{
|
| 415 |
+
"cell_type": "code",
|
| 416 |
+
"execution_count": null,
|
| 417 |
+
"metadata": {},
|
| 418 |
+
"outputs": [],
|
| 419 |
+
"source": [
|
| 420 |
+
"import os\n",
|
| 421 |
+
"os.chdir('../../..')\n",
|
| 422 |
+
"!pip install -e .\n",
|
| 423 |
+
"import sys\n",
|
| 424 |
+
"sys.path.append('fuson_plm')\n",
|
| 425 |
+
"os.chdir('fuson_plm/benchmarking/caid')"
|
| 426 |
+
]
|
| 427 |
+
},
|
| 428 |
+
{
|
| 429 |
+
"cell_type": "code",
|
| 430 |
+
"execution_count": 16,
|
| 431 |
+
"metadata": {},
|
| 432 |
+
"outputs": [],
|
| 433 |
+
"source": [
|
| 434 |
+
"# Metrics calculation\n",
|
| 435 |
+
"def calculate_metrics(preds, labels, threshold=0.5):\n",
|
| 436 |
+
" \"\"\"\n",
|
| 437 |
+
" Calculates metrics to assess model performance\n",
|
| 438 |
+
" Args:\n",
|
| 439 |
+
" preds (list): model's predictions (probabilities)\n",
|
| 440 |
+
" labels (list): ground truth labels\n",
|
| 441 |
+
" threshold (float): minimum threshold a prediction must be met to be considered disordered\n",
|
| 442 |
+
" Returns:\n",
|
| 443 |
+
" accuracy (float): accuracy\n",
|
| 444 |
+
" precision (float): precision\n",
|
| 445 |
+
" recall (float): recall\n",
|
| 446 |
+
" f1 (float): F1 score\n",
|
| 447 |
+
" roc_auc (float): AUROC score\n",
|
| 448 |
+
" \"\"\"\n",
|
| 449 |
+
" flat_binary_preds, flat_prob_preds, flat_labels = [], [], []\n",
|
| 450 |
+
"\n",
|
| 451 |
+
" for pred, label in zip(preds, labels):\n",
|
| 452 |
+
" flat_binary_preds.extend((pred > threshold).astype(int).flatten()) # binary preds are 1 or 0; 1 if the prob > threshold\n",
|
| 453 |
+
" flat_prob_preds.extend(pred.flatten())\n",
|
| 454 |
+
" flat_labels.extend(label.flatten())\n",
|
| 455 |
+
"\n",
|
| 456 |
+
" flat_binary_preds = np.array(flat_binary_preds)\n",
|
| 457 |
+
" flat_prob_preds = np.array(flat_prob_preds)\n",
|
| 458 |
+
" flat_labels = np.array(flat_labels)\n",
|
| 459 |
+
"\n",
|
| 460 |
+
" accuracy = accuracy_score(flat_labels, flat_binary_preds)\n",
|
| 461 |
+
" precision = precision_score(flat_labels, flat_binary_preds)\n",
|
| 462 |
+
" recall = recall_score(flat_labels, flat_binary_preds)\n",
|
| 463 |
+
" f1 = f1_score(flat_labels, flat_binary_preds)\n",
|
| 464 |
+
" roc_auc = roc_auc_score(flat_labels, flat_prob_preds)\n",
|
| 465 |
+
" \n",
|
| 466 |
+
" # make dictionary of the results and return it\n",
|
| 467 |
+
" metrics_dict = {\n",
|
| 468 |
+
" 'Accuracy': accuracy, \n",
|
| 469 |
+
" 'Precision': precision, \n",
|
| 470 |
+
" 'Recall': recall, \n",
|
| 471 |
+
" 'F1 Score': f1, \n",
|
| 472 |
+
" 'AUROC': roc_auc\n",
|
| 473 |
+
" }\n",
|
| 474 |
+
"\n",
|
| 475 |
+
" return metrics_dict"
|
| 476 |
+
]
|
| 477 |
+
},
|
| 478 |
+
{
|
| 479 |
+
"cell_type": "code",
|
| 480 |
+
"execution_count": null,
|
| 481 |
+
"metadata": {},
|
| 482 |
+
"outputs": [],
|
| 483 |
+
"source": [
|
| 484 |
+
"# Recalculate the true statistics\n",
|
| 485 |
+
"prob_and_label_df = model_predictions_labeled[['prob_1','Label']]\n",
|
| 486 |
+
"probs = ','.join(prob_and_label_df['prob_1'].tolist())\n",
|
| 487 |
+
"probs = [float(x) for x in probs.split(\",\")]\n",
|
| 488 |
+
"true_labels = ''.join(prob_and_label_df['Label'].tolist())\n",
|
| 489 |
+
"true_labels = [int(x) for x in list(true_labels)]\n",
|
| 490 |
+
"\n",
|
| 491 |
+
"calculate_metrics(np.array(probs), np.array(true_labels), threshold=0.022)"
|
| 492 |
+
]
|
| 493 |
+
},
|
| 494 |
+
{
|
| 495 |
+
"cell_type": "markdown",
|
| 496 |
+
"metadata": {
|
| 497 |
+
"id": "w25hagtZaV65"
|
| 498 |
+
},
|
| 499 |
+
"source": [
|
| 500 |
+
"### Visualize on Favorite Fusions"
|
| 501 |
+
]
|
| 502 |
+
},
|
| 503 |
+
{
|
| 504 |
+
"cell_type": "code",
|
| 505 |
+
"execution_count": null,
|
| 506 |
+
"metadata": {},
|
| 507 |
+
"outputs": [],
|
| 508 |
+
"source": [
|
| 509 |
+
"l = model_predictions_labeled['FusionGene'].tolist()\n",
|
| 510 |
+
"for i in range(len(l)):\n",
|
| 511 |
+
" print(i, l[i])"
|
| 512 |
+
]
|
| 513 |
+
},
|
| 514 |
+
{
|
| 515 |
+
"cell_type": "code",
|
| 516 |
+
"execution_count": 21,
|
| 517 |
+
"metadata": {},
|
| 518 |
+
"outputs": [],
|
| 519 |
+
"source": [
|
| 520 |
+
"def visualize_fusion_protein_fourgrid(selected_row, save_normalized_propensities=False):\n",
|
| 521 |
+
" cif_file = f\"{fusion_structure_folder}/{selected_row['Fusion_Structure_Link']}\"\n",
|
| 522 |
+
" plddt_values = [float(x) for x in selected_row['Fusion_AA_pLDDTs'].split(\",\")]\n",
|
| 523 |
+
" disorder_values = [float(x) for x in selected_row['prob_1'].split(\",\")]\n",
|
| 524 |
+
" head_indices = selected_row[\"top_hg_UniProt_fus_indices\"]\n",
|
| 525 |
+
" if len(head_indices)>0:\n",
|
| 526 |
+
" head_indices = [int(x) for x in head_indices.split(',')]\n",
|
| 527 |
+
" else:\n",
|
| 528 |
+
" head_indices = []\n",
|
| 529 |
+
" tail_indices = selected_row[\"top_tg_UniProt_fus_indices\"]\n",
|
| 530 |
+
" if len(tail_indices)>0:\n",
|
| 531 |
+
" tail_indices = [int(x) for x in tail_indices.split(',')]\n",
|
| 532 |
+
" else:\n",
|
| 533 |
+
" tail_indices = []\n",
|
| 534 |
+
" print(f\"Visualizing file: {cif_file}\")\n",
|
| 535 |
+
" print(f\"Fusion protein: {selected_row['FusionGene']}\")\n",
|
| 536 |
+
" print(f\"Fusion pLDDT: {selected_row['Fusion_pLDDT']}\")\n",
|
| 537 |
+
" print(f\"Fusion pcnt_disordered: {selected_row['pcnt_disordered']}\\tpredicted pcnt_disordered: {selected_row['pred_pcnt_disordered']}\")\n",
|
| 538 |
+
" print(f\"Sequence start: {selected_row['sequence'][0:10]}...\")\n",
|
| 539 |
+
" print(f\"Sequence length: {len(selected_row['sequence'])}...\")\n",
|
| 540 |
+
" print(f\"seq_id: {len(selected_row['seq_id'])}...\")\n",
|
| 541 |
+
" print(f\"Fusion indices of best head alignment: {head_indices}\")\n",
|
| 542 |
+
" print(f\"Fusion indices of best tail alignment: {tail_indices}\")\n",
|
| 543 |
+
" print(f\"Disorder values start: {disorder_values[0:10]}...\")\n",
|
| 544 |
+
" \n",
|
| 545 |
+
" print(f\"\\nQuadrants:\\n\\t(0,0): pLDDT values in AF2 coloring\\n\\t(0,1): normalized 100-pLDDT values in AF2 coloring\\n\\t(1,0): head and tail segment coloring\\n\\t(1, 1): FusOn-pLM Disorder Model predictions, normalized, in AF2 coloring\")\n",
|
| 546 |
+
"\n",
|
| 547 |
+
" #color_by_head_tail(cif_file, head_indices, tail_indices)\n",
|
| 548 |
+
" #color_by_pLDDT(cif_file, plddt_values)\n",
|
| 549 |
+
" four_coloring_viewer_grid(cif_file, plddt_values, disorder_values, head_indices, tail_indices, \n",
|
| 550 |
+
" seq_id=selected_row['seq_id'],\n",
|
| 551 |
+
" save_normalized_propensities=save_normalized_propensities)"
|
| 552 |
+
]
|
| 553 |
+
},
|
| 554 |
+
{
|
| 555 |
+
"cell_type": "code",
|
| 556 |
+
"execution_count": null,
|
| 557 |
+
"metadata": {},
|
| 558 |
+
"outputs": [],
|
| 559 |
+
"source": [
|
| 560 |
+
"selected_row = model_predictions_labeled.iloc[103,:]\n",
|
| 561 |
+
"visualize_fusion_protein_fourgrid(selected_row,save_normalized_propensities=True)"
|
| 562 |
+
]
|
| 563 |
+
},
|
| 564 |
+
{
|
| 565 |
+
"cell_type": "code",
|
| 566 |
+
"execution_count": null,
|
| 567 |
+
"metadata": {
|
| 568 |
+
"colab": {
|
| 569 |
+
"base_uri": "https://localhost:8080/",
|
| 570 |
+
"height": 497
|
| 571 |
+
},
|
| 572 |
+
"id": "Vr9ZECa8sMUg",
|
| 573 |
+
"outputId": "30b38391-9482-4cd5-8997-bb245fe5e6e5"
|
| 574 |
+
},
|
| 575 |
+
"outputs": [],
|
| 576 |
+
"source": [
|
| 577 |
+
"# Example usage:\n",
|
| 578 |
+
"### Used for FIgure 4D \n",
|
| 579 |
+
"selected_row = model_predictions_labeled.iloc[103,:]\n",
|
| 580 |
+
"cif_file = f\"{fusion_structure_folder}/{selected_row['Fusion_Structure_Link']}\"\n",
|
| 581 |
+
"disorder_values = [float(x) for x in selected_row['prob_1'].split(\",\")]\n",
|
| 582 |
+
"print(f\"Visualizing file: {cif_file}\")\n",
|
| 583 |
+
"print(f\"Fusion protein: {selected_row['FusionGene']}\")\n",
|
| 584 |
+
"print(f\"Fusion pLDDT: {selected_row['Fusion_pLDDT']}\")\n",
|
| 585 |
+
"print(f\"Fusion pcnt_disordered: {selected_row['pcnt_disordered']}\\tpredicted pcnt_disordered: {selected_row['pred_pcnt_disordered']}\")\n",
|
| 586 |
+
"print(f\"Sequence start: {selected_row['sequence'][0:10]}...\")\n",
|
| 587 |
+
"print(f\"Sequence length: {len(selected_row['sequence'])}...\")\n",
|
| 588 |
+
"print(f\"Disorder values start: {disorder_values[0:10]}...\")\n",
|
| 589 |
+
"color_by_disorder(cif_file, disorder_values)"
|
| 590 |
+
]
|
| 591 |
+
},
|
| 592 |
+
{
|
| 593 |
+
"cell_type": "code",
|
| 594 |
+
"execution_count": null,
|
| 595 |
+
"metadata": {},
|
| 596 |
+
"outputs": [],
|
| 597 |
+
"source": [
|
| 598 |
+
"#### Used for disorder figure\n",
|
| 599 |
+
"selected_row = model_predictions_labeled.loc[\n",
|
| 600 |
+
" model_predictions_labeled['FusionGene'].str.contains('EWSR1::FLI1')\n",
|
| 601 |
+
"].reset_index(drop=True).iloc[0,:]\n",
|
| 602 |
+
"\n",
|
| 603 |
+
"visualize_fusion_protein_fourgrid(selected_row,save_normalized_propensities=True)"
|
| 604 |
+
]
|
| 605 |
+
},
|
| 606 |
+
{
|
| 607 |
+
"cell_type": "code",
|
| 608 |
+
"execution_count": null,
|
| 609 |
+
"metadata": {},
|
| 610 |
+
"outputs": [],
|
| 611 |
+
"source": [
|
| 612 |
+
"selected_row = model_predictions_labeled.loc[\n",
|
| 613 |
+
" model_predictions_labeled['FusionGene'].str.contains('BCR::ABL')\n",
|
| 614 |
+
"].reset_index(drop=True).iloc[-1,:]\n",
|
| 615 |
+
"\n",
|
| 616 |
+
"visualize_fusion_protein_fourgrid(selected_row,save_normalized_propensities=True)\n",
|
| 617 |
+
"cif_file = cif_file = f\"{fusion_structure_folder}/{selected_row['Fusion_Structure_Link']}\"\n",
|
| 618 |
+
"disorder_values = [float(x) for x in selected_row['prob_1'].split(\",\")]\n",
|
| 619 |
+
"color_by_disorder(cif_file, disorder_values)\n"
|
| 620 |
+
]
|
| 621 |
+
},
|
| 622 |
+
{
|
| 623 |
+
"cell_type": "code",
|
| 624 |
+
"execution_count": null,
|
| 625 |
+
"metadata": {},
|
| 626 |
+
"outputs": [],
|
| 627 |
+
"source": [
|
| 628 |
+
"selected_row = model_predictions_labeled.loc[\n",
|
| 629 |
+
" model_predictions_labeled['FusionGene'].str.contains('EML4::ALK')\n",
|
| 630 |
+
"].reset_index(drop=True).iloc[0,:]\n",
|
| 631 |
+
"\n",
|
| 632 |
+
"visualize_fusion_protein_fourgrid(selected_row,save_normalized_propensities=True)\n",
|
| 633 |
+
"cif_file = f\"{fusion_structure_folder}/{selected_row['Fusion_Structure_Link']}\"\n",
|
| 634 |
+
"disorder_values = [float(x) for x in selected_row['prob_1'].split(\",\")]\n",
|
| 635 |
+
"color_by_disorder(cif_file, disorder_values)\n"
|
| 636 |
+
]
|
| 637 |
+
},
|
| 638 |
+
{
|
| 639 |
+
"cell_type": "code",
|
| 640 |
+
"execution_count": null,
|
| 641 |
+
"metadata": {},
|
| 642 |
+
"outputs": [],
|
| 643 |
+
"source": [
|
| 644 |
+
"selected_row = model_predictions_labeled.loc[\n",
|
| 645 |
+
" model_predictions_labeled['FusionGene'].str.contains('SS18::SSX1')\n",
|
| 646 |
+
"].reset_index(drop=True).iloc[0,:]\n",
|
| 647 |
+
"\n",
|
| 648 |
+
"visualize_fusion_protein_fourgrid(selected_row,save_normalized_propensities=True)\n",
|
| 649 |
+
"cif_file = f\"{fusion_structure_folder}/{selected_row['Fusion_Structure_Link']}\"\n",
|
| 650 |
+
"disorder_values = [float(x) for x in selected_row['prob_1'].split(\",\")]\n",
|
| 651 |
+
"color_by_disorder(cif_file, disorder_values)\n"
|
| 652 |
+
]
|
| 653 |
+
},
|
| 654 |
+
{
|
| 655 |
+
"cell_type": "markdown",
|
| 656 |
+
"metadata": {},
|
| 657 |
+
"source": [
|
| 658 |
+
"# Visualize important fusions, colored by head/tail and by actual pLDDT"
|
| 659 |
+
]
|
| 660 |
+
},
|
| 661 |
+
{
|
| 662 |
+
"cell_type": "code",
|
| 663 |
+
"execution_count": 28,
|
| 664 |
+
"metadata": {},
|
| 665 |
+
"outputs": [],
|
| 666 |
+
"source": [
|
| 667 |
+
"def visualize_fusion_protein_twogrid(selected_row):\n",
|
| 668 |
+
" cif_file = f\"{fusion_structure_folder}/{selected_row['Fusion_Structure_Link']}\"\n",
|
| 669 |
+
" plddt_values = [float(x) for x in selected_row['Fusion_AA_pLDDTs'].split(\",\")]\n",
|
| 670 |
+
" head_indices = selected_row[\"top_hg_UniProt_fus_indices\"]\n",
|
| 671 |
+
" if len(head_indices)>0:\n",
|
| 672 |
+
" head_indices = [int(x) for x in head_indices.split(',')]\n",
|
| 673 |
+
" else:\n",
|
| 674 |
+
" head_indices = []\n",
|
| 675 |
+
" tail_indices = selected_row[\"top_tg_UniProt_fus_indices\"]\n",
|
| 676 |
+
" if len(tail_indices)>0:\n",
|
| 677 |
+
" tail_indices = [int(x) for x in tail_indices.split(',')]\n",
|
| 678 |
+
" else:\n",
|
| 679 |
+
" tail_indices = []\n",
|
| 680 |
+
" print(f\"Visualizing file: {cif_file}\")\n",
|
| 681 |
+
" print(f\"Fusion protein: {selected_row['FusionGene']}\")\n",
|
| 682 |
+
" print(f\"Fusion pLDDT: {selected_row['Fusion_pLDDT']}\")\n",
|
| 683 |
+
" print(f\"Sequence start: {selected_row['Fusion_Seq'][0:10]}...\")\n",
|
| 684 |
+
" print(f\"Sequence length: {len(selected_row['Fusion_Seq'])}...\")\n",
|
| 685 |
+
" print(f\"Fusion indices of best head alignment: {head_indices}\")\n",
|
| 686 |
+
" print(f\"Fusion indices of best tail alignment: {tail_indices}\")\n",
|
| 687 |
+
" #print(f\"Disorder values start: {disorder_values[0:10]}...\")\n",
|
| 688 |
+
"\n",
|
| 689 |
+
" #color_by_head_tail(cif_file, head_indices, tail_indices)\n",
|
| 690 |
+
" #color_by_pLDDT(cif_file, plddt_values)\n",
|
| 691 |
+
" two_coloring_viewer_grid(cif_file, plddt_values, head_indices, tail_indices)"
|
| 692 |
+
]
|
| 693 |
+
},
|
| 694 |
+
{
|
| 695 |
+
"cell_type": "code",
|
| 696 |
+
"execution_count": null,
|
| 697 |
+
"metadata": {},
|
| 698 |
+
"outputs": [],
|
| 699 |
+
"source": [
|
| 700 |
+
"df = pd.read_csv('../../data/fuson_db.csv')\n",
|
| 701 |
+
"df.loc[df['fusiongenes'].str.contains('EML4::ALK')]"
|
| 702 |
+
]
|
| 703 |
+
},
|
| 704 |
+
{
|
| 705 |
+
"cell_type": "code",
|
| 706 |
+
"execution_count": null,
|
| 707 |
+
"metadata": {},
|
| 708 |
+
"outputs": [],
|
| 709 |
+
"source": [
|
| 710 |
+
"fusion_structure_data.loc[\n",
|
| 711 |
+
" fusion_structure_data['FusionGene'].str.contains('PAX3::FOXO1')\n",
|
| 712 |
+
"]"
|
| 713 |
+
]
|
| 714 |
+
},
|
| 715 |
+
{
|
| 716 |
+
"cell_type": "code",
|
| 717 |
+
"execution_count": null,
|
| 718 |
+
"metadata": {},
|
| 719 |
+
"outputs": [],
|
| 720 |
+
"source": [
|
| 721 |
+
"def apply_thresh(y):\n",
|
| 722 |
+
" if y < 68.8:\n",
|
| 723 |
+
" return 1 #disorder\n",
|
| 724 |
+
" return 0\n",
|
| 725 |
+
"\n",
|
| 726 |
+
"fusion_structure_data['n_disorder'] = fusion_structure_data['Fusion_AA_pLDDTs'].apply(lambda x: x.split(','))\n",
|
| 727 |
+
"fusion_structure_data['n_disorder'] = fusion_structure_data['n_disorder'].apply(lambda x: [float(y) for y in x])\n",
|
| 728 |
+
"#68.8\n",
|
| 729 |
+
"fusion_structure_data['n_disorder'] = fusion_structure_data['n_disorder'].apply(lambda x: [apply_thresh(y) for y in x])\n",
|
| 730 |
+
"fusion_structure_data['n_disorder'] = fusion_structure_data['n_disorder'].apply(lambda x: sum(x))\n",
|
| 731 |
+
"fusion_structure_data['pcnt_disorder'] = round(100*fusion_structure_data['n_disorder']/fusion_structure_data['Fusion_Length'],2)\n",
|
| 732 |
+
"fusion_structure_data['pcnt_disorder'] "
|
| 733 |
+
]
|
| 734 |
+
},
|
| 735 |
+
{
|
| 736 |
+
"cell_type": "code",
|
| 737 |
+
"execution_count": 40,
|
| 738 |
+
"metadata": {},
|
| 739 |
+
"outputs": [],
|
| 740 |
+
"source": [
|
| 741 |
+
"os.makedirs(\"disorder_coloring_data\",exist_ok=True)\n",
|
| 742 |
+
"fusion_structure_data.loc[\n",
|
| 743 |
+
" fusion_structure_data['FusionGene'].str.contains('PAX3::FOXO1') |\n",
|
| 744 |
+
" fusion_structure_data['FusionGene'].str.contains('EWSR1::FLI1') |\n",
|
| 745 |
+
" fusion_structure_data['FusionGene'].str.contains('EML4::ALK')|\n",
|
| 746 |
+
" fusion_structure_data['FusionGene'].str.contains('SS18::SSX1')\n",
|
| 747 |
+
"][['FusionGID','FusionGene','pcnt_disorder','Fusion_Length','Fusion_pLDDT']].reset_index(drop=True)#.to_csv(\"disorder_coloring_data/favorite_fusions_all_structures.csv\",index=False)"
|
| 748 |
+
]
|
| 749 |
+
},
|
| 750 |
+
{
|
| 751 |
+
"cell_type": "code",
|
| 752 |
+
"execution_count": null,
|
| 753 |
+
"metadata": {},
|
| 754 |
+
"outputs": [],
|
| 755 |
+
"source": [
|
| 756 |
+
"selected_row = fusion_structure_data.loc[\n",
|
| 757 |
+
" fusion_structure_data['FusionGene'].str.contains('PAX3::FOXO1')\n",
|
| 758 |
+
"].sort_values(by='Fusion_pLDDT',ascending=True).reset_index(drop=True).iloc[0,:]\n",
|
| 759 |
+
"print('GID', selected_row['FusionGID'])\n",
|
| 760 |
+
"print('percent disorder', selected_row['pcnt_disorder'])\n",
|
| 761 |
+
"visualize_fusion_protein_twogrid(selected_row)"
|
| 762 |
+
]
|
| 763 |
+
},
|
| 764 |
+
{
|
| 765 |
+
"cell_type": "code",
|
| 766 |
+
"execution_count": null,
|
| 767 |
+
"metadata": {},
|
| 768 |
+
"outputs": [],
|
| 769 |
+
"source": [
|
| 770 |
+
"selected_row = fusion_structure_data.loc[\n",
|
| 771 |
+
" fusion_structure_data['FusionGene'].str.contains('EWSR1::FLI1')\n",
|
| 772 |
+
"].sort_values(by='Fusion_pLDDT',ascending=True).reset_index(drop=True).iloc[0,:]\n",
|
| 773 |
+
"\n",
|
| 774 |
+
"visualize_fusion_protein_twogrid(selected_row)"
|
| 775 |
+
]
|
| 776 |
+
},
|
| 777 |
+
{
|
| 778 |
+
"cell_type": "code",
|
| 779 |
+
"execution_count": null,
|
| 780 |
+
"metadata": {},
|
| 781 |
+
"outputs": [],
|
| 782 |
+
"source": [
|
| 783 |
+
"selected_row = fusion_structure_data.loc[\n",
|
| 784 |
+
" fusion_structure_data['FusionGene'].str.contains('EML4::ALK')\n",
|
| 785 |
+
"].sort_values(by='Fusion_pLDDT',ascending=True).reset_index(drop=True).iloc[0,:]\n",
|
| 786 |
+
"\n",
|
| 787 |
+
"#visualize_fusion_protein(selected_row)\n",
|
| 788 |
+
"#selected_row[['Fusion_Length','top_hg_UniProt_fus_indices','top_tg_UniProt_fus_indices']]\n",
|
| 789 |
+
"visualize_fusion_protein_twogrid(selected_row)"
|
| 790 |
+
]
|
| 791 |
+
},
|
| 792 |
+
{
|
| 793 |
+
"cell_type": "code",
|
| 794 |
+
"execution_count": null,
|
| 795 |
+
"metadata": {},
|
| 796 |
+
"outputs": [],
|
| 797 |
+
"source": [
|
| 798 |
+
"selected_row = fusion_structure_data.loc[\n",
|
| 799 |
+
" fusion_structure_data['FusionGene'].str.contains('SS18::SSX1')\n",
|
| 800 |
+
"].sort_values(by='Fusion_pLDDT',ascending=True).reset_index(drop=True).iloc[1,:]\n",
|
| 801 |
+
"\n",
|
| 802 |
+
"visualize_fusion_protein_twogrid(selected_row)\n",
|
| 803 |
+
"#selected_row[['Fusion_Length','top_hg_UniProt_fus_indices','top_tg_UniProt_fus_indices']]"
|
| 804 |
+
]
|
| 805 |
+
},
|
| 806 |
+
{
|
| 807 |
+
"cell_type": "code",
|
| 808 |
+
"execution_count": null,
|
| 809 |
+
"metadata": {},
|
| 810 |
+
"outputs": [],
|
| 811 |
+
"source": [
|
| 812 |
+
"model_predictions_labeled.loc[\n",
|
| 813 |
+
" model_predictions_labeled['seq_id'].isin(['seq26176','seq12668','seq4546','seq11520','seq36572'])\n",
|
| 814 |
+
"][['seq_id','FusionGene']]"
|
| 815 |
+
]
|
| 816 |
+
}
|
| 817 |
+
],
|
| 818 |
+
"metadata": {
|
| 819 |
+
"colab": {
|
| 820 |
+
"collapsed_sections": [
|
| 821 |
+
"FJd6a9gdZNjG",
|
| 822 |
+
"zORkLJztZWp9",
|
| 823 |
+
"w25hagtZaV65",
|
| 824 |
+
"IbyqxlvAFUAK",
|
| 825 |
+
"0n5PSprbhLk7"
|
| 826 |
+
],
|
| 827 |
+
"machine_shape": "hm",
|
| 828 |
+
"provenance": []
|
| 829 |
+
},
|
| 830 |
+
"kernelspec": {
|
| 831 |
+
"display_name": "Python 3",
|
| 832 |
+
"name": "python3"
|
| 833 |
+
},
|
| 834 |
+
"language_info": {
|
| 835 |
+
"codemirror_mode": {
|
| 836 |
+
"name": "ipython",
|
| 837 |
+
"version": 3
|
| 838 |
+
},
|
| 839 |
+
"file_extension": ".py",
|
| 840 |
+
"mimetype": "text/x-python",
|
| 841 |
+
"name": "python",
|
| 842 |
+
"nbconvert_exporter": "python",
|
| 843 |
+
"pygments_lexer": "ipython3",
|
| 844 |
+
"version": "3.10.12"
|
| 845 |
+
}
|
| 846 |
+
},
|
| 847 |
+
"nbformat": 4,
|
| 848 |
+
"nbformat_minor": 0
|
| 849 |
+
}
|
fuson_plm/benchmarking/caid/config.py
ADDED
|
@@ -0,0 +1,14 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
# Which models to benchmark
|
| 2 |
+
BENCHMARK_FUSONPLM = True
|
| 3 |
+
# FUSONPLM_CKPTS. If you've traiend your own model, this is a dictionary: key = run name, values = epochs
|
| 4 |
+
# If you want to use the trained FusOn-pLM, instead FUSONPLM_CKPTS="FusOn-pLM"
|
| 5 |
+
FUSONPLM_CKPTS= "FusOn-pLM"
|
| 6 |
+
|
| 7 |
+
BENCHMARK_ESM = True
|
| 8 |
+
|
| 9 |
+
# GPU configs
|
| 10 |
+
CUDA_VISIBLE_DEVICES="0"
|
| 11 |
+
|
| 12 |
+
# Overwriting configs
|
| 13 |
+
PERMISSION_TO_OVERWRITE_EMBEDDINGS = False # if False, script will halt if it believes these embeddings have already been made.
|
| 14 |
+
PERMISSION_TO_OVERWRITE_MODELS = False # if False, script will halt if it believes these embeddings have already been made.
|
fuson_plm/benchmarking/caid/disorder_coloring_data/normalized_disorder_propensities_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:4874f8b7e228a71038c231eca02abf06cee45f63f9bed106f5561b5cfa45c3b3
|
| 3 |
+
size 43282
|
fuson_plm/benchmarking/caid/model.py
ADDED
|
@@ -0,0 +1,26 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
import torch.nn as nn
|
| 2 |
+
|
| 3 |
+
# Transformer model class
|
| 4 |
+
class DisorderPredictor(nn.Module):
|
| 5 |
+
def __init__(self, input_dim, hidden_dim, num_heads, num_layers, dropout):
|
| 6 |
+
super(DisorderPredictor, self).__init__()
|
| 7 |
+
self.embedding_dim = input_dim
|
| 8 |
+
self.self_attention = nn.MultiheadAttention(embed_dim=input_dim, num_heads=num_heads, dropout=dropout)
|
| 9 |
+
encoder_layer = nn.TransformerEncoderLayer(
|
| 10 |
+
d_model=hidden_dim,
|
| 11 |
+
nhead=num_heads,
|
| 12 |
+
dropout=dropout,
|
| 13 |
+
batch_first=True
|
| 14 |
+
)
|
| 15 |
+
self.transformer_encoder = nn.TransformerEncoder(encoder_layer, num_layers=num_layers)
|
| 16 |
+
self.classifier = nn.Linear(input_dim, 1) # juts produce probabilities for 1 class
|
| 17 |
+
#self.softmax = nn.Softmax(dim=-1)
|
| 18 |
+
self.sigmoid = nn.Sigmoid()
|
| 19 |
+
|
| 20 |
+
def forward(self, embeddings):
|
| 21 |
+
attn_out, _ = self.self_attention(embeddings, embeddings, embeddings) # Start with embeddings as random Q, K, V vectors
|
| 22 |
+
transformer_out = self.transformer_encoder(attn_out) # Get outputs from encoder layers
|
| 23 |
+
logits = self.classifier(transformer_out) # Linear classifier
|
| 24 |
+
probs = self.sigmoid(logits.squeeze(-1)) # sigmoid for probabilities; remove the last dimension of size 1 (since we only predicted 1 class)
|
| 25 |
+
return probs # Get probabilities of dimension seq_len
|
| 26 |
+
|
fuson_plm/benchmarking/caid/plot.py
ADDED
|
@@ -0,0 +1,1030 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
import matplotlib.pyplot as plt
|
| 2 |
+
import seaborn as sns
|
| 3 |
+
import pandas as pd
|
| 4 |
+
import numpy as np
|
| 5 |
+
import os
|
| 6 |
+
import matplotlib.colors as mcolors
|
| 7 |
+
import matplotlib.patches as mpatches
|
| 8 |
+
from matplotlib import font_manager
|
| 9 |
+
import matplotlib.patches as patches
|
| 10 |
+
from sklearn.metrics import roc_curve, auc, r2_score
|
| 11 |
+
|
| 12 |
+
from fuson_plm.utils.visualizing import set_font
|
| 13 |
+
|
| 14 |
+
global caid2_winners, caid2_model_rankings
|
| 15 |
+
caid2_winners = pd.DataFrame(data=
|
| 16 |
+
{
|
| 17 |
+
'Model Name': ['Dispredict3','flDPnn2','flDPnn','flDPlr','flDPlr2','DisoPred',
|
| 18 |
+
'IDP-Fusion','ESpritz-D','DeepIDP-2L','disomine','DISOPRED3-diso','IUPred3',
|
| 19 |
+
'AlphaFold-rsa','AlphaFold-pLDDT'], # do the top 6 models, and IUPred because it's well-known
|
| 20 |
+
'AUROC': [0.838,0.836,0.833,0.827,0.821,0.821,
|
| 21 |
+
0.818,0.802,0.800,0.797,0.692,0.755,0.747,0.695],
|
| 22 |
+
})
|
| 23 |
+
caid2_winners['Model Type'] = ['caid2_competition']*len(caid2_winners)
|
| 24 |
+
caid2_winners['Model Epoch'] = [np.nan]*len(caid2_winners)
|
| 25 |
+
|
| 26 |
+
caid2_model_rankings = {
|
| 27 |
+
'Dispredict3': 1,
|
| 28 |
+
'flDPnn2': 2,
|
| 29 |
+
'flDPnn': 3,
|
| 30 |
+
'flDPlr': 4,
|
| 31 |
+
'flDPlr2': 5,
|
| 32 |
+
'DisoPred': 6,
|
| 33 |
+
'IDP-Fusion': 7,
|
| 34 |
+
'ESpritz-D': 8,
|
| 35 |
+
'DeepIDP-2L': 9,
|
| 36 |
+
'disomine': 10,
|
| 37 |
+
'DISOPRED3-diso': 35,
|
| 38 |
+
'IUPred3': 21,
|
| 39 |
+
'AlphaFold-rsa': 24,
|
| 40 |
+
'AlphaFold-pLDDT': 34
|
| 41 |
+
}
|
| 42 |
+
|
| 43 |
+
# Method for lengthening the model name
|
| 44 |
+
def lengthen_model_name(row):
|
| 45 |
+
model_type = row['Model Type']
|
| 46 |
+
name = row['Model Name']
|
| 47 |
+
epoch = row['Model Epoch']
|
| 48 |
+
|
| 49 |
+
if 'esm' in name:
|
| 50 |
+
return name
|
| 51 |
+
if 'puncta' in name:
|
| 52 |
+
return name
|
| 53 |
+
if model_type=='caid2_competition':
|
| 54 |
+
return name
|
| 55 |
+
|
| 56 |
+
return f'{name}_e{epoch}'
|
| 57 |
+
|
| 58 |
+
# Method for shortening the model name for display
|
| 59 |
+
def shorten_model_name(row):
|
| 60 |
+
model_type = row['Model Type']
|
| 61 |
+
name = row['Model Name']
|
| 62 |
+
epoch = row['Model Epoch']
|
| 63 |
+
|
| 64 |
+
if 'esm' in name:
|
| 65 |
+
return 'ESM-2-650M'
|
| 66 |
+
if model_type=='caid2_competition':
|
| 67 |
+
return name
|
| 68 |
+
|
| 69 |
+
if 'snp_' in name:
|
| 70 |
+
prob_type = 'snp'
|
| 71 |
+
elif 'uniform_' in name:
|
| 72 |
+
prob_type = 'uni'
|
| 73 |
+
|
| 74 |
+
layers = name.split('layers')[0].split('_')[-1]
|
| 75 |
+
maskrate = name.split('mask')[1].split('-', 1)[0]
|
| 76 |
+
kqv_tag = name.split('layers_')[1].split('_')[0]
|
| 77 |
+
dt = name.split('mask')[1].split('-', 1)[1]
|
| 78 |
+
|
| 79 |
+
return f'{prob_type}_{layers}L_{kqv_tag}_mask{maskrate}_{dt}_e{epoch}'
|
| 80 |
+
|
| 81 |
+
def make_heatmap(df, results_dir='.', gold_standard_model_name="esm2_t33_650M_UR50D",split="test",thresh=None,ax=None):
|
| 82 |
+
# Set font to Ubuntu
|
| 83 |
+
set_font()
|
| 84 |
+
|
| 85 |
+
# Declare columns to compare: metrics
|
| 86 |
+
columns_to_compare = ['AUROC']
|
| 87 |
+
|
| 88 |
+
# Define the literature-reported values for CAID competition winners - only IF the split is not "benchmark"
|
| 89 |
+
if not(split=="benchmark"):
|
| 90 |
+
df = pd.concat([df,caid2_winners])
|
| 91 |
+
|
| 92 |
+
# Create Short Model Name and Full Model Name columns for later use
|
| 93 |
+
df['Model Epoch'] = df['Model Epoch'].apply(lambda x: str(int(x)) if not(np.isnan(x)) else '')
|
| 94 |
+
df['Short Model Name'] = df.apply(lambda row: shorten_model_name(row),axis=1)
|
| 95 |
+
df['Full Model Name'] = df.apply(lambda row: lengthen_model_name(row), axis=1)
|
| 96 |
+
|
| 97 |
+
# Isolate gold standard row for later comparison
|
| 98 |
+
gold_standard = df[df['Full Model Name'] == gold_standard_model_name].reset_index(drop=True).iloc[0]
|
| 99 |
+
gold_standard_short_model_name = df[df['Full Model Name'] == gold_standard_model_name]['Short Model Name'].item()
|
| 100 |
+
|
| 101 |
+
# Create a new dataframe for the heatmap; sort by model type and place gold standard on top
|
| 102 |
+
heatmap_data = df[['Model Type','Short Model Name','Full Model Name'] + columns_to_compare].copy()
|
| 103 |
+
heatmap_data['is_gold_standard'] = (heatmap_data['Full Model Name'] == gold_standard_model_name).astype(int)
|
| 104 |
+
heatmap_data = heatmap_data.sort_values(by=['is_gold_standard','Model Type','AUROC'], ascending=[False,True,False]).reset_index(drop=True).drop(columns=['is_gold_standard'])
|
| 105 |
+
# Save the original values before calculating differences so we can use them for annotation
|
| 106 |
+
original_values = heatmap_data[columns_to_compare].copy()
|
| 107 |
+
|
| 108 |
+
# Calculate differences from the gold standard
|
| 109 |
+
for col in columns_to_compare:
|
| 110 |
+
heatmap_data[col] = heatmap_data[col] - gold_standard[col]
|
| 111 |
+
|
| 112 |
+
# Create a color map where values equal to 0 are white, above are red, and below are blue
|
| 113 |
+
cmap = sns.color_palette("coolwarm", as_cmap=True) # other option is diverging_palette(220, 20, as_cmap=True)
|
| 114 |
+
|
| 115 |
+
### Make the plot
|
| 116 |
+
# can plot on a bigger plot, or make it an individual plot
|
| 117 |
+
if ax is None:
|
| 118 |
+
tallsize = max(8, 8 +.25*(len(heatmap_data)-26))
|
| 119 |
+
fig, ax = plt.subplots(1, 1, figsize=(8, tallsize), dpi=300)
|
| 120 |
+
|
| 121 |
+
# Plot the heatmap with original values as annotations
|
| 122 |
+
hm = sns.heatmap(heatmap_data.set_index('Short Model Name').drop(columns=['Model Type','Full Model Name']),
|
| 123 |
+
annot=False, fmt='', cmap=cmap, center=0,
|
| 124 |
+
cbar_kws={'label': 'Difference from Gold Standard'})
|
| 125 |
+
|
| 126 |
+
# Explicitly set tick labels to prevent them from being messed up
|
| 127 |
+
ax.set_yticklabels(heatmap_data['Short Model Name'], rotation=0, fontsize=12)
|
| 128 |
+
# Add padding to the y-axis label
|
| 129 |
+
ax.set_ylabel("Short Model Name", labelpad=20) # Increase the labelpad value to add more padding
|
| 130 |
+
|
| 131 |
+
# Bold any values values that exceed the gold standard
|
| 132 |
+
for i in range(original_values.shape[0]):
|
| 133 |
+
for j in range(original_values.shape[1]):
|
| 134 |
+
value = original_values.iloc[i, j]
|
| 135 |
+
if value > gold_standard[columns_to_compare[j]]:
|
| 136 |
+
ax.text(j + 0.5, i + 0.5, f'{value:.3f}', ha='center', va='center', fontweight='bold', color='black')
|
| 137 |
+
else:
|
| 138 |
+
ax.text(j + 0.5, i + 0.5, f'{value:.3f}', ha='center', va='center', color='black')
|
| 139 |
+
|
| 140 |
+
# Add horizontal lines between different model types
|
| 141 |
+
model_type_series = heatmap_data['Model Type'].values
|
| 142 |
+
last_index = 0
|
| 143 |
+
labels_positions = [] # To store the positions for labels
|
| 144 |
+
for i in range(1, len(model_type_series)):
|
| 145 |
+
if model_type_series[i] != model_type_series[i - 1]:
|
| 146 |
+
hm.axhline(i, color='white', linewidth=8) # Draw a thick white line between groups
|
| 147 |
+
labels_positions.append((last_index + i) / 2) # Store the midpoint for labeling
|
| 148 |
+
last_index = i
|
| 149 |
+
|
| 150 |
+
# Add label for the last group
|
| 151 |
+
labels_positions.append((last_index + len(model_type_series)) / 2)
|
| 152 |
+
|
| 153 |
+
# Italic and bold models that win AUROC; apply yellow coloring to gold standard model
|
| 154 |
+
for ytick, model_name in enumerate(heatmap_data['Short Model Name']):
|
| 155 |
+
if model_name == gold_standard_short_model_name:
|
| 156 |
+
# color yellow
|
| 157 |
+
label = ax.get_yticklabels()[ytick]
|
| 158 |
+
#label.set_color('gold')
|
| 159 |
+
label.set_bbox(dict(facecolor='gold', alpha=0.5, edgecolor='gold'))
|
| 160 |
+
if model_name != gold_standard_short_model_name:
|
| 161 |
+
auroc_value = original_values.loc[ytick, 'AUROC']
|
| 162 |
+
|
| 163 |
+
# Apply bold and italic for wins on either AUROC or F1 Score
|
| 164 |
+
if (auroc_value > gold_standard['AUROC']):
|
| 165 |
+
label = ax.get_yticklabels()[ytick]
|
| 166 |
+
#label.set_style('italic')
|
| 167 |
+
#label.set_weight('bold')
|
| 168 |
+
label.set_bbox(dict(facecolor='red', alpha=0.3, edgecolor='red'))
|
| 169 |
+
|
| 170 |
+
# Make legend
|
| 171 |
+
gold_patch = mpatches.Patch(color='gold', alpha=0.5, label='Gold Standard')
|
| 172 |
+
red_patch = mpatches.Patch(color='red', alpha=0.5, label='Winner')
|
| 173 |
+
plt.legend(handles=[gold_patch, red_patch], loc='best', bbox_to_anchor=(0, 0)) # You can change loc to position the legend
|
| 174 |
+
|
| 175 |
+
split_fname_dict = {
|
| 176 |
+
"testing": "CAID2_test",
|
| 177 |
+
"training": "CAID2_train",
|
| 178 |
+
"benchmark": "FusionPDB_pLDDT_disorder"
|
| 179 |
+
}
|
| 180 |
+
split_title_dict = {
|
| 181 |
+
"testing": "CAID-2 Disorder Prediction",
|
| 182 |
+
"training": "CAID-2 Disorder Prediction",
|
| 183 |
+
"benchmark": "FusionPDB_pLDDT Disorder Prediction"
|
| 184 |
+
}
|
| 185 |
+
ax.set_title(split_title_dict[split])
|
| 186 |
+
|
| 187 |
+
# Rotate the color bar label
|
| 188 |
+
cbar = hm.collections[0].colorbar
|
| 189 |
+
cbar.ax.yaxis.set_label_position('right')
|
| 190 |
+
cbar.ax.yaxis.set_ticks_position('right')
|
| 191 |
+
cbar.set_label('Difference from Gold Standard', rotation=270, labelpad=20) # Rotate 270 degrees and add some padding
|
| 192 |
+
|
| 193 |
+
# Set tight layout using fig
|
| 194 |
+
fig.tight_layout(rect=[0, 0, 0.95, 1]) # Add extra padding on the right side to fit the label
|
| 195 |
+
|
| 196 |
+
plt.savefig(f"{results_dir}/{split_fname_dict[split]}_heatmap_vs_{gold_standard_model_name}.png")
|
| 197 |
+
|
| 198 |
+
# Plot AUROC curve of ONE model of interest on its fusion pdb performance
|
| 199 |
+
def make_benchmark_auroc_curve(results_dir='.', seq_label_dict=None, path_to_results_of_interest='', model_alias=None):
|
| 200 |
+
# Isolate the information for the model we'll be plotting
|
| 201 |
+
benchmark_model = path_to_results_of_interest.split('trained_models/')[1].split('/')
|
| 202 |
+
benchmark_model_type = benchmark_model[0]
|
| 203 |
+
benchmark_model_epoch = np.nan
|
| 204 |
+
benchmark_model_hyperparams = None
|
| 205 |
+
if len(benchmark_model)==5:
|
| 206 |
+
benchmark_model_name = benchmark_model[1]
|
| 207 |
+
benchmark_model_epoch = benchmark_model[2].split('epoch')[1]
|
| 208 |
+
benchmark_model_hyperparams = benchmark_model[3]
|
| 209 |
+
else:
|
| 210 |
+
benchmark_model_name = benchmark_model[0]
|
| 211 |
+
benchmark_model_hyperparams = benchmark_model[1]
|
| 212 |
+
benchmark_model_info = pd.DataFrame(data={
|
| 213 |
+
'Model Type': [benchmark_model_type], 'Model Name': [benchmark_model_name], 'Model Epoch': [benchmark_model_epoch]
|
| 214 |
+
})
|
| 215 |
+
if model_alias is None:
|
| 216 |
+
model_alias = benchmark_model_info.apply(lambda row: shorten_model_name(row),axis=1).iloc[0]
|
| 217 |
+
|
| 218 |
+
color_map = {
|
| 219 |
+
model_alias: 'black'
|
| 220 |
+
}
|
| 221 |
+
method_results = {model_alias: path_to_results_of_interest}
|
| 222 |
+
method_results = {k:v for k,v in method_results.items() if v not in [None, '']}
|
| 223 |
+
|
| 224 |
+
set_font()
|
| 225 |
+
plt.figure(figsize=(10,6),dpi=300)
|
| 226 |
+
|
| 227 |
+
# To store AUROC values and corresponding labels for sorting
|
| 228 |
+
roc_data = []
|
| 229 |
+
# Read each result file and plot the metrics
|
| 230 |
+
for method, path in method_results.items():
|
| 231 |
+
df = pd.read_csv(path) # columns = prob_1,labels
|
| 232 |
+
|
| 233 |
+
# Extract probabilities and labels
|
| 234 |
+
prob_1 = ",".join(df['prob_1'].tolist())
|
| 235 |
+
df['labels'] = df['sequence'].apply(lambda x: seq_label_dict[x])
|
| 236 |
+
labels = "".join(df['labels'].tolist())
|
| 237 |
+
prob_1 = [float(x) for x in prob_1.split(",")]
|
| 238 |
+
labels = [int(x) for x in list(labels)]
|
| 239 |
+
sequences = "".join(df['sequence'].tolist())
|
| 240 |
+
assert len(prob_1)==len(labels)==len(sequences)
|
| 241 |
+
|
| 242 |
+
# Compute ROC curve and ROC area
|
| 243 |
+
fpr, tpr, thresholds = roc_curve(labels, prob_1)
|
| 244 |
+
roc_auc = auc(fpr, tpr)
|
| 245 |
+
|
| 246 |
+
# Store data for sorting later
|
| 247 |
+
roc_data.append((method, fpr, tpr, roc_auc))
|
| 248 |
+
|
| 249 |
+
# Sort the methods by AUROC values
|
| 250 |
+
roc_data = sorted(roc_data, key=lambda x: x[3], reverse=True)
|
| 251 |
+
|
| 252 |
+
# Plot sorted ROC curves
|
| 253 |
+
for method, fpr, tpr, roc_auc in roc_data:
|
| 254 |
+
if method == model_alias:
|
| 255 |
+
plt.plot(fpr, tpr, color=color_map[method], lw=2, label=f'{method} ({roc_auc:0.3f})')
|
| 256 |
+
else:
|
| 257 |
+
plt.plot(fpr, tpr, color=color_map[method], lw=1, alpha=0.7, label=f'{method} ({roc_auc:0.3f})')
|
| 258 |
+
|
| 259 |
+
# Set other stylistic elements
|
| 260 |
+
plt.xlim([0.0, 1.0])
|
| 261 |
+
plt.ylim([0.0, 1.05])
|
| 262 |
+
plt.plot([0, 1], [0, 1], color='darkgrey', lw=2, linestyle='--')
|
| 263 |
+
plt.xlabel('False Positive Rate')
|
| 264 |
+
plt.ylabel('True Positive Rate')
|
| 265 |
+
plt.title('Receiver Operating Characteristic (ROC) Curve')
|
| 266 |
+
|
| 267 |
+
# After plotting the ROC curves, customize the legend
|
| 268 |
+
handles, labels = plt.gca().get_legend_handles_labels()
|
| 269 |
+
|
| 270 |
+
# Create the legend first
|
| 271 |
+
legend = plt.legend(handles, labels, loc="center left", bbox_to_anchor=(1, 0.5))
|
| 272 |
+
|
| 273 |
+
# Iterate through the legend's text labels
|
| 274 |
+
for text in legend.get_texts():
|
| 275 |
+
if model_alias in text.get_text():
|
| 276 |
+
text.set_fontweight('bold') # Bold the alias model
|
| 277 |
+
|
| 278 |
+
plt.tight_layout()
|
| 279 |
+
plt.savefig(f'{results_dir}/FusionPDB_pLDDT_disorder_{model_alias}_AUROC_curve.png')
|
| 280 |
+
|
| 281 |
+
# Plot AUROC curve of ONE model of interest with all the CAID models
|
| 282 |
+
def make_auroc_curve(results_dir='.', seq_label_dict=None, seq_ids_dict=None, path_to_results_of_interest='', model_alias=None, path_to_esm_results=None, with_rankings=False):
|
| 283 |
+
# Isolate the information for the model we'll be plotting
|
| 284 |
+
benchmark_model = path_to_results_of_interest.split('trained_models/')[1].split('/')
|
| 285 |
+
benchmark_model_type = benchmark_model[0]
|
| 286 |
+
benchmark_model_epoch = np.nan
|
| 287 |
+
benchmark_model_hyperparams = None
|
| 288 |
+
if len(benchmark_model)==5:
|
| 289 |
+
benchmark_model_name = benchmark_model[1]
|
| 290 |
+
benchmark_model_epoch = benchmark_model[2].split('epoch')[1]
|
| 291 |
+
benchmark_model_hyperparams = benchmark_model[3]
|
| 292 |
+
else:
|
| 293 |
+
benchmark_model_name = benchmark_model[0]
|
| 294 |
+
benchmark_model_hyperparams = benchmark_model[1]
|
| 295 |
+
benchmark_model_info = pd.DataFrame(data={
|
| 296 |
+
'Model Type': [benchmark_model_type], 'Model Name': [benchmark_model_name], 'Model Epoch': [benchmark_model_epoch]
|
| 297 |
+
})
|
| 298 |
+
if model_alias is None:
|
| 299 |
+
model_alias = benchmark_model_info.apply(lambda row: shorten_model_name(row),axis=1).iloc[0]
|
| 300 |
+
|
| 301 |
+
color_map = {
|
| 302 |
+
'Dispredict3': '#d62727', #1
|
| 303 |
+
'flDPnn2': '#ff7f0f', #2
|
| 304 |
+
'flDPnn': '#1f77b4', #3
|
| 305 |
+
'flDPlr': '#bcbd21', #4
|
| 306 |
+
'flDPlr2': '#16becf', #5
|
| 307 |
+
'DisoPred': '#1f77b4', #6
|
| 308 |
+
'IDP-Fusion': '#d62727', #7
|
| 309 |
+
'ESpritz-D': '#8b564c', #8
|
| 310 |
+
'DeepIDP-2L': '#e377c2', #9
|
| 311 |
+
'disomine': '#e377c2', #10
|
| 312 |
+
'DISOPRED3-diso': '#ff892d',
|
| 313 |
+
'IUPred3': '#8b564c',
|
| 314 |
+
'AlphaFold-rsa': '#2ba02b',
|
| 315 |
+
'AlphaFold-pLDDT': '#ff892d',
|
| 316 |
+
model_alias: 'black'
|
| 317 |
+
}
|
| 318 |
+
method_results = {'Dispredict3': 'processed_data/caid2_competition_results/Dispredict3_CAID-2_Disorder_NOX.csv',
|
| 319 |
+
'flDPnn2': 'processed_data/caid2_competition_results/flDPnn2_CAID-2_Disorder_NOX.csv',
|
| 320 |
+
'flDPnn': 'processed_data/caid2_competition_results/flDPnn_CAID-2_Disorder_NOX.csv',
|
| 321 |
+
'flDPlr': 'processed_data/caid2_competition_results/flDPtr_CAID-2_Disorder_NOX.csv', # name doesn't match but this is what it is in raw download
|
| 322 |
+
'flDPlr2': 'processed_data/caid2_competition_results/flDPlr2_CAID-2_Disorder_NOX.csv',
|
| 323 |
+
'DisoPred': 'processed_data/caid2_competition_results/DisoPred_CAID-2_Disorder_NOX.csv',
|
| 324 |
+
'IDP-Fusion': 'processed_data/caid2_competition_results/IDP-Fusion_CAID-2_Disorder_NOX.csv',
|
| 325 |
+
'ESpritz-D': 'processed_data/caid2_competition_results/ESpritz-D_CAID-2_Disorder_NOX.csv',
|
| 326 |
+
'DeepIDP-2L': 'processed_data/caid2_competition_results/DeepIDP-2L_CAID-2_Disorder_NOX.csv',
|
| 327 |
+
'disomine': 'processed_data/caid2_competition_results/disomine_CAID-2_Disorder_NOX.csv',
|
| 328 |
+
'DISOPRED3-diso': 'processed_data/caid2_competition_results/DISOPRED3-diso_CAID-2_Disorder_NOX.csv',
|
| 329 |
+
'AlphaFold-rsa': 'processed_data/caid2_competition_results/AlphaFold-rsa_CAID-2_Disorder_NOX.csv',
|
| 330 |
+
'AlphaFold-pLDDT': 'processed_data/caid2_competition_results/AlphaFold-disorder_CAID-2_Disorder_NOX.csv', # name doesn't match but this is what it is in raw download
|
| 331 |
+
'IUPred3': 'processed_data/caid2_competition_results/IUPred3_CAID-2_Disorder_NOX.csv',
|
| 332 |
+
model_alias: path_to_results_of_interest
|
| 333 |
+
}
|
| 334 |
+
if path_to_esm_results is not None:
|
| 335 |
+
method_results['ESM-2-650M'] = path_to_esm_results
|
| 336 |
+
color_map['ESM-2-650M'] = 'black'
|
| 337 |
+
|
| 338 |
+
method_results = {k:v for k,v in method_results.items() if v not in [None, '']}
|
| 339 |
+
|
| 340 |
+
set_font()
|
| 341 |
+
plt.figure(figsize=(12,6),dpi=300)
|
| 342 |
+
|
| 343 |
+
# To store AUROC values and corresponding labels for sorting
|
| 344 |
+
merged_preds = pd.DataFrame(data={'sequence':[]})
|
| 345 |
+
merged_tpr_fpr = pd.DataFrame(data={'model': [],'fpr':[],'tpr':[]})
|
| 346 |
+
roc_data = []
|
| 347 |
+
# Read each result file and plot the metrics
|
| 348 |
+
for method, path in method_results.items():
|
| 349 |
+
df = pd.read_csv(path) # columns = prob_1,labels
|
| 350 |
+
merged_preds = pd.merge(merged_preds,
|
| 351 |
+
df.rename(columns={'prob_1':f"{method}_prob_1"})[['sequence',f"{method}_prob_1",]],
|
| 352 |
+
on=['sequence'],how='outer')
|
| 353 |
+
|
| 354 |
+
# Extract probabilities and labels
|
| 355 |
+
prob_1 = ",".join(df['prob_1'].tolist())
|
| 356 |
+
df['labels'] = df['sequence'].apply(lambda x: seq_label_dict[x])
|
| 357 |
+
labels = "".join(df['labels'].tolist())
|
| 358 |
+
prob_1 = [float(x) for x in prob_1.split(",")]
|
| 359 |
+
labels = [int(x) for x in list(labels)]
|
| 360 |
+
sequences = "".join(df['sequence'].tolist())
|
| 361 |
+
assert len(prob_1)==len(labels)==len(sequences)
|
| 362 |
+
|
| 363 |
+
# Compute ROC curve and ROC area
|
| 364 |
+
fpr, tpr, thresholds = roc_curve(labels, prob_1)
|
| 365 |
+
new_tpr_fpr = pd.DataFrame(data={
|
| 366 |
+
'model': [method]*len(fpr),
|
| 367 |
+
'fpr': fpr, 'tpr': tpr
|
| 368 |
+
})
|
| 369 |
+
merged_tpr_fpr = pd.concat([merged_tpr_fpr,new_tpr_fpr])
|
| 370 |
+
roc_auc = auc(fpr, tpr)
|
| 371 |
+
|
| 372 |
+
if method==model_alias:
|
| 373 |
+
path_to_og_metrics = path_to_results_of_interest.rsplit('/',1)[0]+'/caid_hyperparam_screen_test_metrics.csv'
|
| 374 |
+
og_metrics = pd.read_csv(path_to_og_metrics)
|
| 375 |
+
roc_auc = og_metrics['AUROC'][0]
|
| 376 |
+
|
| 377 |
+
# Store data for sorting later
|
| 378 |
+
roc_data.append((method, fpr, tpr, roc_auc))
|
| 379 |
+
|
| 380 |
+
# Save the merged dataframe as source data
|
| 381 |
+
merged_preds['labels'] = merged_preds['sequence'].apply(lambda x: seq_label_dict[x])
|
| 382 |
+
merged_preds['labels'] = merged_preds['labels'].apply(lambda x: ",".join([str(y) for y in x]))
|
| 383 |
+
merged_preds['ids'] = merged_preds['sequence'].apply(lambda x: seq_ids_dict[x])
|
| 384 |
+
merged_preds.drop(columns={'sequence'}).to_csv(f"{results_dir}/CAID_prediction_source_data.csv",index=False)
|
| 385 |
+
merged_tpr_fpr.to_csv(f"{results_dir}/CAID_fpr_tpr_source_data.csv",index=False)
|
| 386 |
+
# Sort the methods by AUROC values
|
| 387 |
+
roc_data = sorted(roc_data, key=lambda x: x[3], reverse=True)
|
| 388 |
+
|
| 389 |
+
# figure out the labels
|
| 390 |
+
labels = {method: method for method in method_results}
|
| 391 |
+
if with_rankings:
|
| 392 |
+
for method in labels:
|
| 393 |
+
if method in caid2_model_rankings:
|
| 394 |
+
labels[method] = f"{caid2_model_rankings[method]}. {method}"
|
| 395 |
+
|
| 396 |
+
# Plot sorted ROC curves
|
| 397 |
+
for method, fpr, tpr, roc_auc in roc_data:
|
| 398 |
+
if method=='ESM-2-650M' and path_to_esm_results is not None:
|
| 399 |
+
plt.plot(fpr, tpr, color=color_map[method], lw=2, linestyle='--', label=f'{labels[method]} ({roc_auc:0.3f})')
|
| 400 |
+
elif method == model_alias:
|
| 401 |
+
plt.plot(fpr, tpr, color=color_map[method], lw=2, label=f'{labels[method]} ({roc_auc:0.3f})')
|
| 402 |
+
else:
|
| 403 |
+
plt.plot(fpr, tpr, color=color_map[method], lw=1, alpha=0.7, label=f'{labels[method]} ({roc_auc:0.3f})')
|
| 404 |
+
|
| 405 |
+
# Set other stylistic elements
|
| 406 |
+
plt.xlim([0.0, 1.0])
|
| 407 |
+
plt.ylim([0.0, 1.05])
|
| 408 |
+
plt.xticks(fontsize=20)
|
| 409 |
+
plt.yticks(fontsize=20)
|
| 410 |
+
plt.plot([0, 1], [0, 1], color='darkgrey', lw=2, linestyle='--')
|
| 411 |
+
plt.xlabel('False Positive Rate', fontsize=22)
|
| 412 |
+
plt.ylabel('True Positive Rate', fontsize=22)
|
| 413 |
+
plt.title('CAID2 Disorder NOX Dataset: ROC Curve', fontsize=22)
|
| 414 |
+
|
| 415 |
+
# After plotting the ROC curves, customize the legend
|
| 416 |
+
handles, labels = plt.gca().get_legend_handles_labels()
|
| 417 |
+
|
| 418 |
+
# Create the legend first
|
| 419 |
+
legend = plt.legend(handles, labels, loc="center left", bbox_to_anchor=(1.1, 0.5), fontsize=16)
|
| 420 |
+
|
| 421 |
+
# Iterate through the legend's text labels
|
| 422 |
+
for text in legend.get_texts():
|
| 423 |
+
if model_alias in text.get_text():
|
| 424 |
+
text.set_fontweight('bold') # Bold the alias model
|
| 425 |
+
elif (path_to_esm_results is not None) and "ESM-2-650M" in text.get_text():
|
| 426 |
+
text.set_fontweight('bold') # Bold ESM if we're comparing to it
|
| 427 |
+
|
| 428 |
+
plt.tight_layout()
|
| 429 |
+
figpath = f'{results_dir}/CAID2_{model_alias}_AUROC_curve.png'
|
| 430 |
+
if path_to_esm_results is not None:
|
| 431 |
+
figpath = f'{results_dir}/CAID2_{model_alias}_with_ESM_AUROC_curve.png'
|
| 432 |
+
plt.savefig(figpath)
|
| 433 |
+
|
| 434 |
+
|
| 435 |
+
def plot_disorder_content_scatter(train_labels, test_labels, benchmark_labels, savepath='splits/disorder_content_scatter.png'):
|
| 436 |
+
"""
|
| 437 |
+
Compare disorder content between the train, test, and fusion benchmark sets based on the TRUE labels.
|
| 438 |
+
Each labels vector should have ['11110000','0001110',...] format.
|
| 439 |
+
"""
|
| 440 |
+
|
| 441 |
+
# Get train disorder distribution
|
| 442 |
+
train_lengths = []
|
| 443 |
+
train_frac_disorder = []
|
| 444 |
+
for vec in train_labels:
|
| 445 |
+
veclist = [int(x) for x in vec]
|
| 446 |
+
train_lengths.append(len(veclist))
|
| 447 |
+
train_frac_disorder.append(sum(veclist)/len(veclist))
|
| 448 |
+
|
| 449 |
+
# Get test disorder distribution
|
| 450 |
+
test_lengths = []
|
| 451 |
+
test_frac_disorder = []
|
| 452 |
+
for vec in test_labels:
|
| 453 |
+
veclist = [int(x) for x in vec]
|
| 454 |
+
test_lengths.append(len(veclist))
|
| 455 |
+
test_frac_disorder.append(sum(veclist)/len(veclist))
|
| 456 |
+
|
| 457 |
+
# Get benchmark disorder distribution
|
| 458 |
+
benchmark_lengths = []
|
| 459 |
+
benchmark_frac_disorder = []
|
| 460 |
+
for vec in benchmark_labels:
|
| 461 |
+
veclist = [int(x) for x in vec]
|
| 462 |
+
benchmark_lengths.append(len(veclist))
|
| 463 |
+
benchmark_frac_disorder.append(sum(veclist)/len(veclist))
|
| 464 |
+
|
| 465 |
+
# make a plot
|
| 466 |
+
set_font()
|
| 467 |
+
color_map = {
|
| 468 |
+
'train': '#0072B2',
|
| 469 |
+
'test': '#E69F00',
|
| 470 |
+
'fusion': 'purple'
|
| 471 |
+
}
|
| 472 |
+
|
| 473 |
+
# Plotting
|
| 474 |
+
fig, ax = plt.subplots(figsize=(10, 6))
|
| 475 |
+
|
| 476 |
+
ax.scatter(train_lengths, train_frac_disorder, color=color_map['train'], label='Train', alpha=0.7)
|
| 477 |
+
ax.scatter(test_lengths, test_frac_disorder, color=color_map['test'], label='Test', alpha=0.7)
|
| 478 |
+
ax.scatter(benchmark_lengths, benchmark_frac_disorder, color=color_map['fusion'], label='Fusion', alpha=0.7)
|
| 479 |
+
|
| 480 |
+
# Labels and title
|
| 481 |
+
ax.set_xlabel('Length')
|
| 482 |
+
ax.set_ylabel('Fraction of Disorder')
|
| 483 |
+
ax.set_title('Length vs. Fraction of Disorder for Train, Test, and Benchmark Datasets')
|
| 484 |
+
ax.legend()
|
| 485 |
+
plt.tight_layout()
|
| 486 |
+
plt.savefig(savepath)
|
| 487 |
+
|
| 488 |
+
def plot_disorder_content_hist(labels, ids, title="data", color="black", savepath='splits/disorder_content_histograms.png'):
|
| 489 |
+
"""
|
| 490 |
+
Compare disorder content between the train, test, and fusion benchmark sets based on the TRUE labels.
|
| 491 |
+
Each labels vector should have ['11110000','0001110',...] format.
|
| 492 |
+
"""
|
| 493 |
+
set_font()
|
| 494 |
+
|
| 495 |
+
# Get disorder distribution
|
| 496 |
+
lengths = []
|
| 497 |
+
frac_disorder = []
|
| 498 |
+
for vec in labels:
|
| 499 |
+
veclist = [int(x) for x in vec]
|
| 500 |
+
lengths.append(len(veclist))
|
| 501 |
+
frac_disorder.append(100*sum(veclist)/len(veclist)) # make it a percent, i like this better
|
| 502 |
+
|
| 503 |
+
# save the source data
|
| 504 |
+
source_data = pd.DataFrame(data={
|
| 505 |
+
'ID': ids,
|
| 506 |
+
'Percent_Disordered': frac_disorder
|
| 507 |
+
})
|
| 508 |
+
source_data['Percent_Disordered'] = source_data['Percent_Disordered'].round(3)
|
| 509 |
+
source_data.to_csv(savepath.replace(".png","_source_data.csv"),index=False)
|
| 510 |
+
|
| 511 |
+
fig, ax = plt.subplots(1, 1, figsize=(20, 12))
|
| 512 |
+
|
| 513 |
+
# Plot histogram for train data
|
| 514 |
+
title_fontsize = 70
|
| 515 |
+
axislabel_fontsize = 70
|
| 516 |
+
tick_fontsize = 50
|
| 517 |
+
ax.hist(frac_disorder, bins=20, color=color, alpha=0.7)
|
| 518 |
+
ax.set_title(title, fontsize=title_fontsize)
|
| 519 |
+
ax.set_xlabel('% Disordered', fontsize=axislabel_fontsize)
|
| 520 |
+
ax.set_ylabel('Count', fontsize=axislabel_fontsize)
|
| 521 |
+
ax.grid(True)
|
| 522 |
+
ax.set_axisbelow(True)
|
| 523 |
+
ax.tick_params(axis='both', which='major', labelsize=tick_fontsize)
|
| 524 |
+
|
| 525 |
+
# Calculate the mean and median of the percent coverage
|
| 526 |
+
mean_coverage = np.mean(frac_disorder)
|
| 527 |
+
median_coverage = np.median(frac_disorder)
|
| 528 |
+
|
| 529 |
+
# Add vertical line for the mean
|
| 530 |
+
ax.axvline(mean_coverage, color='black', linestyle='--', linewidth=2, label=f'Mean: {mean_coverage:.1f}%')
|
| 531 |
+
|
| 532 |
+
# Add vertical line for the median
|
| 533 |
+
ax.axvline(median_coverage, color='black', linestyle='-', linewidth=2, label=f'Median: {median_coverage:.1f}%')
|
| 534 |
+
|
| 535 |
+
ax.legend(fontsize=50, title_fontsize=50)
|
| 536 |
+
|
| 537 |
+
plt.tight_layout()
|
| 538 |
+
plt.savefig(savepath)
|
| 539 |
+
|
| 540 |
+
def plot_group_disorder_content_hist(train_labels, test_labels, benchmark_labels, savepath='splits/disorder_content_histograms.png',orient='horizontal'):
|
| 541 |
+
"""
|
| 542 |
+
Compare disorder content between the train, test, and fusion benchmark sets based on the TRUE labels.
|
| 543 |
+
Each labels vector should have ['11110000','0001110',...] format.
|
| 544 |
+
"""
|
| 545 |
+
|
| 546 |
+
# Get train disorder distribution
|
| 547 |
+
train_lengths = []
|
| 548 |
+
train_frac_disorder = []
|
| 549 |
+
for vec in train_labels:
|
| 550 |
+
veclist = [int(x) for x in vec]
|
| 551 |
+
train_lengths.append(len(veclist))
|
| 552 |
+
train_frac_disorder.append(sum(veclist)/len(veclist))
|
| 553 |
+
|
| 554 |
+
# Get test disorder distribution
|
| 555 |
+
test_lengths = []
|
| 556 |
+
test_frac_disorder = []
|
| 557 |
+
for vec in test_labels:
|
| 558 |
+
veclist = [int(x) for x in vec]
|
| 559 |
+
test_lengths.append(len(veclist))
|
| 560 |
+
test_frac_disorder.append(sum(veclist)/len(veclist))
|
| 561 |
+
|
| 562 |
+
# Get benchmark disorder distribution
|
| 563 |
+
benchmark_lengths = []
|
| 564 |
+
benchmark_frac_disorder = []
|
| 565 |
+
for vec in benchmark_labels:
|
| 566 |
+
veclist = [int(x) for x in vec]
|
| 567 |
+
benchmark_lengths.append(len(veclist))
|
| 568 |
+
benchmark_frac_disorder.append(sum(veclist)/len(veclist))
|
| 569 |
+
|
| 570 |
+
# make a plot
|
| 571 |
+
set_font()
|
| 572 |
+
color_map = {
|
| 573 |
+
'train': '#0072B2',
|
| 574 |
+
'test': '#E69F00',
|
| 575 |
+
'fusion': 'mediumpurple'
|
| 576 |
+
}
|
| 577 |
+
|
| 578 |
+
# Create a 1x3 subplot (1 row, 3 columns) or 3x1
|
| 579 |
+
if orient=='horizontal':
|
| 580 |
+
fig, axes = plt.subplots(1, 3, figsize=(15, 5), sharey=False)
|
| 581 |
+
if orient=='vertical':
|
| 582 |
+
fig, axes = plt.subplots(3, 1, figsize=(5, 15), sharey=False)
|
| 583 |
+
|
| 584 |
+
# Plot histogram for train data
|
| 585 |
+
title_fontsize = 26
|
| 586 |
+
axislabel_fontsize = 26
|
| 587 |
+
tick_fontsize = 16
|
| 588 |
+
axes[0].hist(train_frac_disorder, bins=20, color=color_map['train'], alpha=0.7)
|
| 589 |
+
axes[0].set_title('CAID2 Train', fontsize=title_fontsize)
|
| 590 |
+
if orient=="horizontal":
|
| 591 |
+
axes[0].set_xlabel('Fraction of Disorder', fontsize=axislabel_fontsize)
|
| 592 |
+
axes[0].set_ylabel('Frequency', fontsize=axislabel_fontsize)
|
| 593 |
+
axes[0].grid(True)
|
| 594 |
+
axes[0].set_axisbelow(True)
|
| 595 |
+
axes[0].tick_params(axis='both', which='major', labelsize=tick_fontsize)
|
| 596 |
+
|
| 597 |
+
|
| 598 |
+
# Plot histogram for test data
|
| 599 |
+
axes[1].hist(test_frac_disorder, bins=20, color=color_map['test'], alpha=0.7)
|
| 600 |
+
axes[1].set_title('CAID2 Test',fontsize=title_fontsize)
|
| 601 |
+
if orient=="horizontal":
|
| 602 |
+
axes[1].set_xlabel('Fraction of Disorder', fontsize=axislabel_fontsize)
|
| 603 |
+
if orient=="vertical":
|
| 604 |
+
axes[1].set_ylabel('Frequency', fontsize=axislabel_fontsize)
|
| 605 |
+
axes[1].grid(True)
|
| 606 |
+
axes[1].set_axisbelow(True)
|
| 607 |
+
axes[1].tick_params(axis='both', which='major', labelsize=tick_fontsize)
|
| 608 |
+
|
| 609 |
+
# Plot histogram for benchmark (fusion) data
|
| 610 |
+
axes[2].hist(benchmark_frac_disorder, bins=20, color=color_map['fusion'], alpha=0.7)
|
| 611 |
+
axes[2].set_title('Fusion Oncoproteins',fontsize=title_fontsize)
|
| 612 |
+
axes[2].set_xlabel('Fraction of Disorder', fontsize=axislabel_fontsize)
|
| 613 |
+
if orient=="vertical":
|
| 614 |
+
axes[2].set_ylabel('Frequency', fontsize=axislabel_fontsize)
|
| 615 |
+
axes[2].grid(True)
|
| 616 |
+
axes[2].set_axisbelow(True)
|
| 617 |
+
axes[2].tick_params(axis='both', which='major', labelsize=tick_fontsize)
|
| 618 |
+
plt.tight_layout()
|
| 619 |
+
plt.savefig(savepath)
|
| 620 |
+
|
| 621 |
+
def categorize_plddt(values):
|
| 622 |
+
categories = {
|
| 623 |
+
"<= 50": sum(1 for x in values if x <= 50),
|
| 624 |
+
"50-70": sum(1 for x in values if 50 < x <= 70),
|
| 625 |
+
"70-90": sum(1 for x in values if 70 < x <= 90),
|
| 626 |
+
"> 90": sum(1 for x in values if x > 90)
|
| 627 |
+
}
|
| 628 |
+
return categories
|
| 629 |
+
|
| 630 |
+
|
| 631 |
+
def plot_fusion_sequence_pLDDT_left_to_right(fusion_structure_data, fusiongene, save_path=''):
|
| 632 |
+
"""
|
| 633 |
+
Plot each amino acid in the sequence as a separate colored bar based on pLDDT values.
|
| 634 |
+
"""
|
| 635 |
+
set_font()
|
| 636 |
+
# Filter for specific fusion data and preprocess
|
| 637 |
+
df_of_interest = fusion_structure_data[fusion_structure_data['FusionGene'] == fusiongene].copy()
|
| 638 |
+
df_of_interest['Fusion_AA_pLDDTs'] = df_of_interest['Fusion_AA_pLDDTs'].apply(lambda x: [float(i) for i in x.split(',')])
|
| 639 |
+
df_of_interest['Label'] = df_of_interest['Fusion_Length'].astype(str) + 'AAs'
|
| 640 |
+
|
| 641 |
+
# Sort data by Fusion_Length
|
| 642 |
+
df_of_interest = df_of_interest.sort_values(by='Fusion_Length', ascending=True).reset_index(drop=True)
|
| 643 |
+
|
| 644 |
+
# Define colors for each pLDDT range
|
| 645 |
+
category_colors = {"<= 50": "#f27842", "50-70": "#f8d514", "70-90": "#60c1e8", "> 90": "#004ecb"}
|
| 646 |
+
|
| 647 |
+
# Helper function to get color based on pLDDT
|
| 648 |
+
def get_color(pLDDT):
|
| 649 |
+
if pLDDT > 90:
|
| 650 |
+
return category_colors["> 90"]
|
| 651 |
+
elif pLDDT > 70:
|
| 652 |
+
return category_colors["70-90"]
|
| 653 |
+
elif pLDDT > 50:
|
| 654 |
+
return category_colors["50-70"]
|
| 655 |
+
else:
|
| 656 |
+
return category_colors["<= 50"]
|
| 657 |
+
|
| 658 |
+
# Start plotting each sequence with colored bars
|
| 659 |
+
fig, ax = plt.subplots(figsize=(10, 6))
|
| 660 |
+
if len(df_of_interest)<3:
|
| 661 |
+
fig, ax = plt.subplots(figsize=(10, 2))
|
| 662 |
+
|
| 663 |
+
average_plddt = dict(zip(df_of_interest['Label'], df_of_interest['Fusion_pLDDT']))
|
| 664 |
+
df_of_interest['Fusion_AA_colors'] = df_of_interest['Fusion_AA_pLDDTs'].apply(lambda x: [get_color(plddt) for plddt in x])
|
| 665 |
+
df_of_interest['Fusion_pLDDT_color'] = df_of_interest['Fusion_pLDDT'].apply(lambda plddt: get_color(plddt))
|
| 666 |
+
# just save the columns needed for the plot
|
| 667 |
+
df_of_interest[['FusionGene','seq_id','Fusion_Length','Fusion_pLDDT','Fusion_AA_pLDDTs','Fusion_AA_colors','Fusion_pLDDT_color',
|
| 668 |
+
'top_hg_UniProtID','top_hg_UniProt_isoform','top_hg_UniProt_fus_indices',
|
| 669 |
+
'top_tg_UniProtID','top_tg_UniProt_isoform','top_tg_UniProt_fus_indices']].to_csv(f"{save_path}/plddt_sequence_{fusiongene}_source_data.csv",index=False)
|
| 670 |
+
|
| 671 |
+
for idx, row in df_of_interest.iterrows():
|
| 672 |
+
pLDDT_values = row['Fusion_AA_pLDDTs']
|
| 673 |
+
colors = [get_color(plddt) for plddt in pLDDT_values]
|
| 674 |
+
|
| 675 |
+
# Plot each amino acid in the sequence with the respective color
|
| 676 |
+
ax.bar(range(len(pLDDT_values)),
|
| 677 |
+
[0.7] * len(pLDDT_values), color=colors, edgecolor='none',
|
| 678 |
+
bottom=idx - 0.7 / 2) # Centering each row at idx
|
| 679 |
+
|
| 680 |
+
labels = df_of_interest['Label'].tolist()
|
| 681 |
+
# Annotate each bar with the Fusion_pLDDT value on the right, colored by PLDDT category
|
| 682 |
+
for idx, label in enumerate(labels):
|
| 683 |
+
avg_plddt_value = average_plddt[label]
|
| 684 |
+
|
| 685 |
+
# Determine color based on the PLDDT category
|
| 686 |
+
if avg_plddt_value > 90:
|
| 687 |
+
color = '#004ecb'
|
| 688 |
+
elif avg_plddt_value > 70:
|
| 689 |
+
color = "#60c1e8"
|
| 690 |
+
elif avg_plddt_value > 50:
|
| 691 |
+
color = '#f8d514'
|
| 692 |
+
else:
|
| 693 |
+
color = '#f27842'
|
| 694 |
+
|
| 695 |
+
# Annotate with the determined color
|
| 696 |
+
if len(df_of_interest)>10:
|
| 697 |
+
markersize = 10
|
| 698 |
+
elif len(df_of_interest)>5:
|
| 699 |
+
markersize = 16
|
| 700 |
+
else:
|
| 701 |
+
markersize=12
|
| 702 |
+
ax.plot(1.02*max(df_of_interest['Fusion_Length']),
|
| 703 |
+
idx, marker='o', color="black", markersize=markersize, markerfacecolor=color, markeredgewidth=2)
|
| 704 |
+
|
| 705 |
+
# Add breakpoint box - make sure we actually HAVE one of each
|
| 706 |
+
hg_indices, tg_indices = None, None
|
| 707 |
+
if not(type(df_of_interest['top_hg_UniProt_fus_indices'][idx])==float):
|
| 708 |
+
hg_indices = [int(x) for x in df_of_interest['top_hg_UniProt_fus_indices'][idx].split(',')]
|
| 709 |
+
if not(type(df_of_interest['top_tg_UniProt_fus_indices'][idx])==float):
|
| 710 |
+
tg_indices = [int(x) for x in df_of_interest['top_tg_UniProt_fus_indices'][idx].split(',')]
|
| 711 |
+
print(hg_indices, tg_indices)
|
| 712 |
+
|
| 713 |
+
if (hg_indices is not None) and (tg_indices is not None):
|
| 714 |
+
box_start = min(hg_indices[-1],tg_indices[0])
|
| 715 |
+
box_end = max(hg_indices[-1],tg_indices[0])
|
| 716 |
+
elif hg_indices is not None:
|
| 717 |
+
box_start, box_end = hg_indices[-1], hg_indices[-1]
|
| 718 |
+
elif tg_indices is not None:
|
| 719 |
+
box_start, box_end = tg_indices[0], tg_indices[0]
|
| 720 |
+
|
| 721 |
+
print(f"box indices for structure {idx}, fusion gene {fusiongene}", box_start, box_end)
|
| 722 |
+
|
| 723 |
+
# Plot the rectangle, making it slightly larger than the rest of the bar
|
| 724 |
+
rect = patches.Rectangle((box_start, idx - 0.7 / 2), box_end-box_start, 0.7, linewidth=2, edgecolor='black', facecolor='none')
|
| 725 |
+
ax.add_patch(rect)
|
| 726 |
+
|
| 727 |
+
# Customize plot
|
| 728 |
+
ax.set_yticks([]) # Hide y-axis ticks
|
| 729 |
+
ax.set_yticklabels([]) # Hide y-axis labels
|
| 730 |
+
ax.set_ylim(-0.5, len(df_of_interest) - 0.5) # reduce white space at top
|
| 731 |
+
ax.set_xlabel("Amino Acid Sequence (ordered)", fontsize=14)
|
| 732 |
+
# Customize x-axis for labeling
|
| 733 |
+
ax.set_xlim(left=0) # Start x-axis at 0 to make bars flush left
|
| 734 |
+
ax.set_xlabel("Amino Acid Sequence (ordered)", fontsize=14)
|
| 735 |
+
ax.tick_params(axis='x', labelsize=30)
|
| 736 |
+
|
| 737 |
+
|
| 738 |
+
plt.title(f"{fusiongene} pLDDT Distribution by Amino Acid Sequence", fontsize=16)
|
| 739 |
+
plt.tight_layout()
|
| 740 |
+
|
| 741 |
+
# Save figure
|
| 742 |
+
fusiongene_savename = fusiongene.replace("::","-")
|
| 743 |
+
plt.savefig(f"{save_path}/plddt_sequence_{fusiongene_savename}.png", dpi=300)
|
| 744 |
+
plt.show()
|
| 745 |
+
|
| 746 |
+
def plot_favorite_fusion_pLDDT_distribution(fusion_structure_data, fusiongene, save_path=''):
|
| 747 |
+
"""
|
| 748 |
+
Make a stacked bar chart of the pLDDT distribution
|
| 749 |
+
"""
|
| 750 |
+
set_font()
|
| 751 |
+
# Filter for EWSR1::FLI1 fusion data and preprocess
|
| 752 |
+
df_of_interest = fusion_structure_data[fusion_structure_data['FusionGene'] == fusiongene].copy()
|
| 753 |
+
df_of_interest['Fusion_AA_pLDDTs'] = df_of_interest['Fusion_AA_pLDDTs'].apply(lambda x: [float(i) for i in x.split(',')])
|
| 754 |
+
df_of_interest['Label'] = df_of_interest['Fusion_Length'].astype(str) + 'AAs'
|
| 755 |
+
# Sort data by Fusion_Length
|
| 756 |
+
df_of_interest = df_of_interest.sort_values(by='Fusion_Length', ascending=True).reset_index(drop=True)
|
| 757 |
+
# Convert to dictionary format
|
| 758 |
+
data_dict = dict(zip(df_of_interest['Label'], df_of_interest['Fusion_AA_pLDDTs']))
|
| 759 |
+
average_plddt = dict(zip(df_of_interest['Label'], df_of_interest['Fusion_pLDDT']))
|
| 760 |
+
|
| 761 |
+
# Categorize each structure
|
| 762 |
+
categorized_data = {structure: categorize_plddt(plddt_values) for structure, plddt_values in data_dict.items()}
|
| 763 |
+
|
| 764 |
+
# Extract counts for each category
|
| 765 |
+
labels = list(categorized_data.keys())
|
| 766 |
+
categories = ["<= 50", "50-70", "70-90", "> 90"]
|
| 767 |
+
counts = {cat: [categorized_data[structure][cat] for structure in labels] for cat in categories}
|
| 768 |
+
|
| 769 |
+
# Define colors for each category
|
| 770 |
+
category_colors = {"<= 50": "#f27842", "50-70": "#f8d514", "70-90": "#60c1e8", "> 90": "#004ecb"}
|
| 771 |
+
|
| 772 |
+
# Re-categorize PLDDT values for the bar chart
|
| 773 |
+
categorized_data = {structure: categorize_plddt(plddt_values) for structure, plddt_values in data_dict.items()}
|
| 774 |
+
labels = list(categorized_data.keys())
|
| 775 |
+
counts = {cat: [categorized_data[structure][cat] for structure in labels] for cat in categories}
|
| 776 |
+
|
| 777 |
+
# Plotting the horizontal stacked bar chart with annotations for 'Fusion_pLDDT' values
|
| 778 |
+
fig, ax = plt.subplots(figsize=(10, 6))
|
| 779 |
+
if len(data_dict)<3:
|
| 780 |
+
fig, ax = plt.subplots(figsize=(10, 2))
|
| 781 |
+
bottom = np.zeros(len(labels))
|
| 782 |
+
|
| 783 |
+
# Stack each category horizontally
|
| 784 |
+
for cat in categories:
|
| 785 |
+
ax.barh(labels, counts[cat], label=cat, color=category_colors[cat], left=bottom)
|
| 786 |
+
bottom += counts[cat] # Update the left position for the next stack
|
| 787 |
+
|
| 788 |
+
# Annotate each bar with the Fusion_pLDDT value on the right, colored by PLDDT category
|
| 789 |
+
for idx, label in enumerate(labels):
|
| 790 |
+
avg_plddt_value = average_plddt[label]
|
| 791 |
+
|
| 792 |
+
# Determine color based on the PLDDT category
|
| 793 |
+
if avg_plddt_value > 90:
|
| 794 |
+
color = '#004ecb'
|
| 795 |
+
elif avg_plddt_value > 70:
|
| 796 |
+
color = "#60c1e8"
|
| 797 |
+
elif avg_plddt_value > 50:
|
| 798 |
+
color = '#f8d514'
|
| 799 |
+
else:
|
| 800 |
+
color = '#f27842'
|
| 801 |
+
|
| 802 |
+
# Annotate with the determined color
|
| 803 |
+
#ax.text(bottom[idx] + 1, idx, f"{avg_plddt_value:.2f}", va='center', ha='left', color="black", fontsize=18, fontweight='bold')
|
| 804 |
+
if len(df_of_interest)>10:
|
| 805 |
+
markersize = 10
|
| 806 |
+
elif len(df_of_interest)>5:
|
| 807 |
+
markersize = 16
|
| 808 |
+
else:
|
| 809 |
+
markersize=12
|
| 810 |
+
ax.plot(bottom[idx] + .02*max(df_of_interest['Fusion_Length']), idx, marker='s', color="black", markersize=markersize, markerfacecolor=color, markeredgewidth=2)
|
| 811 |
+
|
| 812 |
+
|
| 813 |
+
# Add labels and legend
|
| 814 |
+
#ax.set_xlim([0,max(df_of_interest['Fusion_Length'])*1.0])
|
| 815 |
+
#ax.set_ylabel("Structures")
|
| 816 |
+
# Save original ticks before changing label size
|
| 817 |
+
#ax.tick_params(axis='x', labelsize=16)
|
| 818 |
+
#original_xticks = ax.get_xticks()
|
| 819 |
+
# Set ticks explicitly to avoid automatic adjustment
|
| 820 |
+
#ax.set_xticks(original_xticks)
|
| 821 |
+
|
| 822 |
+
#ax.set_xlabel("Length",fontsize=40)
|
| 823 |
+
ax.tick_params(axis='x', labelsize=30)
|
| 824 |
+
#ax.tick_params(axis='y', labelsize=16)
|
| 825 |
+
ax.tick_params(axis='y', left=False, labelleft=False)
|
| 826 |
+
#ax.set_title(f"{fusiongene} pLDDT Distribution")
|
| 827 |
+
#ax.legend(title="pLDDT Ranges", fontsize=16, bbox_to_anchor=(1, 1), title_fontsize=16)
|
| 828 |
+
|
| 829 |
+
plt.tight_layout()
|
| 830 |
+
fusiongene_savename = fusiongene.replace("::","-")
|
| 831 |
+
plt.savefig(f"{save_path}/plddt_dist_{fusiongene_savename}.png",dpi=300)
|
| 832 |
+
|
| 833 |
+
def make_all_favorite_fusion_pLDDT_plots(favorite_fusions,left_to_right=True):
|
| 834 |
+
fusion_structure_data = pd.read_csv('processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv')
|
| 835 |
+
swissprot_top_alignments = pd.read_csv("../../data/blast/blast_outputs/swissprot_top_alignments.csv")
|
| 836 |
+
fuson_db = pd.read_csv("../../data/fuson_db.csv")
|
| 837 |
+
seq_id_dict = dict(zip(fuson_db['aa_seq'],fuson_db['seq_id']))
|
| 838 |
+
fusion_structure_data['seq_id'] = fusion_structure_data['Fusion_Seq'].map(seq_id_dict)
|
| 839 |
+
fusion_structure_data = pd.merge(
|
| 840 |
+
fusion_structure_data,
|
| 841 |
+
swissprot_top_alignments,
|
| 842 |
+
on="seq_id",
|
| 843 |
+
how="left"
|
| 844 |
+
)
|
| 845 |
+
for x in favorite_fusions:
|
| 846 |
+
if left_to_right:
|
| 847 |
+
plot_fusion_sequence_pLDDT_left_to_right(fusion_structure_data, x, save_path='processed_data/figures/fusion_disorder')
|
| 848 |
+
else:
|
| 849 |
+
plot_favorite_fusion_pLDDT_distribution(fusion_structure_data, x, save_path='processed_data/figures/fusion_disorder')
|
| 850 |
+
|
| 851 |
+
def prep_data_for_ht_disorder_comparison():
|
| 852 |
+
ht_structure_data = pd.read_csv('processed_data/fusionpdb/heads_tails_structural_data.csv')
|
| 853 |
+
fusion_structure_data = pd.read_csv('processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv')
|
| 854 |
+
fusion_heads_and_tails = pd.read_csv('processed_data/fusionpdb/fusion_heads_and_tails.csv')
|
| 855 |
+
|
| 856 |
+
all_hts_with_structures = ht_structure_data['UniProtID'].unique().tolist()
|
| 857 |
+
|
| 858 |
+
fuson_ht_db = pd.read_csv('../../data/blast/fuson_ht_db.csv')[['seq_id','aa_seq','fusiongenes','hgUniProt','tgUniProt']]
|
| 859 |
+
|
| 860 |
+
merge = pd.merge(
|
| 861 |
+
fuson_ht_db.rename(columns={'aa_seq':'Fusion_Seq'}),
|
| 862 |
+
fusion_structure_data[['FusionGID', 'Fusion_Seq','Fusion_pLDDT','Fusion_AA_pLDDTs']],
|
| 863 |
+
on='Fusion_Seq',
|
| 864 |
+
how='right'
|
| 865 |
+
)
|
| 866 |
+
# now merge again
|
| 867 |
+
merge['hgUniProt'] = merge['hgUniProt'].apply(lambda x: x.split(','))
|
| 868 |
+
merge['tgUniProt'] = merge['tgUniProt'].apply(lambda x: x.split(','))
|
| 869 |
+
merge = merge.explode('hgUniProt')
|
| 870 |
+
merge = merge.explode('tgUniProt')
|
| 871 |
+
merge = merge.loc[
|
| 872 |
+
merge['hgUniProt'].isin(all_hts_with_structures) &
|
| 873 |
+
merge['tgUniProt'].isin(all_hts_with_structures)
|
| 874 |
+
].reset_index(drop=True)
|
| 875 |
+
|
| 876 |
+
merge = pd.merge(
|
| 877 |
+
merge,
|
| 878 |
+
ht_structure_data.rename(columns=
|
| 879 |
+
{'UniProtID':'hgUniProt',
|
| 880 |
+
'Avg pLDDT': 'hg_pLDDT',
|
| 881 |
+
'All pLDDTs': 'hg_AA_pLDDTs',
|
| 882 |
+
'Seq': 'hg_seq'}),
|
| 883 |
+
on='hgUniProt',
|
| 884 |
+
how='inner'
|
| 885 |
+
)
|
| 886 |
+
|
| 887 |
+
merge = pd.merge(
|
| 888 |
+
merge,
|
| 889 |
+
ht_structure_data.rename(columns=
|
| 890 |
+
{'UniProtID':'tgUniProt',
|
| 891 |
+
'Avg pLDDT': 'tg_pLDDT',
|
| 892 |
+
'All pLDDTs': 'tg_AA_pLDDTs',
|
| 893 |
+
'Seq': 'tg_seq'}),
|
| 894 |
+
on='tgUniProt',
|
| 895 |
+
how='inner'
|
| 896 |
+
)
|
| 897 |
+
merge = merge.loc[merge['hg_AA_pLDDTs'].notna()]
|
| 898 |
+
merge = merge.loc[merge['tg_AA_pLDDTs'].notna()].reset_index(drop=True)
|
| 899 |
+
|
| 900 |
+
# finally, calcualte label
|
| 901 |
+
merge['hg_label'] = merge['hg_AA_pLDDTs'].apply(lambda x: x.split(','))
|
| 902 |
+
merge['hg_label'] = merge['hg_label'].apply(lambda x: [float(y) for y in x])
|
| 903 |
+
merge['hg_label'] = merge['hg_label'].apply(lambda x: [apply_plddt_thresh(y) for y in x])
|
| 904 |
+
merge['hg_label'] = merge['hg_label'].apply(lambda x: ''.join(x))
|
| 905 |
+
|
| 906 |
+
merge['tg_label'] = merge['tg_AA_pLDDTs'].apply(lambda x: x.split(','))
|
| 907 |
+
merge['tg_label'] = merge['tg_label'].apply(lambda x: [float(y) for y in x])
|
| 908 |
+
merge['tg_label'] = merge['tg_label'].apply(lambda x: [apply_plddt_thresh(y) for y in x])
|
| 909 |
+
merge['tg_label'] = merge['tg_label'].apply(lambda x: ''.join(x))
|
| 910 |
+
|
| 911 |
+
merge['fusion_label'] = merge['Fusion_AA_pLDDTs'].apply(lambda x: x.split(','))
|
| 912 |
+
merge['fusion_label'] = merge['fusion_label'].apply(lambda x: [float(y) for y in x])
|
| 913 |
+
merge['fusion_label'] = merge['fusion_label'].apply(lambda x: [apply_plddt_thresh(y) for y in x])
|
| 914 |
+
merge['fusion_label'] = merge['fusion_label'].apply(lambda x: ''.join(x))
|
| 915 |
+
|
| 916 |
+
return merge
|
| 917 |
+
|
| 918 |
+
def apply_plddt_thresh(y):
|
| 919 |
+
if y < 68.8:
|
| 920 |
+
return '1'
|
| 921 |
+
else:
|
| 922 |
+
return '0'
|
| 923 |
+
|
| 924 |
+
def plot_fusion_stats_boxplots(data, save_path="fusion_disorder_boxplots.png"):
|
| 925 |
+
set_font()
|
| 926 |
+
# Create box plots
|
| 927 |
+
plt.figure(figsize=(6, 5))
|
| 928 |
+
# for ones that are 100% disordered, AUROC was NaN, so drop these
|
| 929 |
+
box = plt.boxplot([data[col].dropna() for col in data.columns], labels=data.columns, patch_artist=True)
|
| 930 |
+
|
| 931 |
+
# Set color of each box plot
|
| 932 |
+
for patch in box['boxes']:
|
| 933 |
+
patch.set_facecolor('#ff68b4')
|
| 934 |
+
patch.set_edgecolor('#ff68b4')
|
| 935 |
+
|
| 936 |
+
# Customize other elements if needed
|
| 937 |
+
#for whisker in box['whiskers']:
|
| 938 |
+
#whisker.set_color('#ff68b4')
|
| 939 |
+
#for cap in box['caps']:
|
| 940 |
+
#cap.set_color('#ff68b4')
|
| 941 |
+
for median in box['medians']:
|
| 942 |
+
median.set_color('black')
|
| 943 |
+
# Add labels and title
|
| 944 |
+
#plt.xlabel('Metrics')
|
| 945 |
+
#plt.ylabel('Values')
|
| 946 |
+
plt.title(f"Per-Residue Disorder (n={len(data)})",fontsize=22)
|
| 947 |
+
plt.xticks(rotation=20,fontsize=22)
|
| 948 |
+
plt.yticks(fontsize=22)
|
| 949 |
+
|
| 950 |
+
# Show plot
|
| 951 |
+
plt.tight_layout()
|
| 952 |
+
plt.show()
|
| 953 |
+
plt.savefig(save_path,dpi=300)
|
| 954 |
+
|
| 955 |
+
def plot_fusion_frac_disorder_r2(actual_values, predicted_values, save_path="fusion_pred_disorder_r2.png"):
|
| 956 |
+
set_font()
|
| 957 |
+
plt.figure(figsize=(6, 6))
|
| 958 |
+
r2 = r2_score(actual_values, predicted_values)
|
| 959 |
+
#sns.kdeplot(actual_values, label="Actual Values", shade=True)
|
| 960 |
+
#sns.kdeplot(predicted_values, label="Predicted Values", shade=True)
|
| 961 |
+
plt.scatter(actual_values, predicted_values, alpha=0.5, label=f"Predictions", color="#ff68b4")
|
| 962 |
+
plt.plot([min(actual_values), max(actual_values)], [min(actual_values), max(actual_values)], 'k--', label='Ideal Fit')
|
| 963 |
+
plt.text(0, 92, f"$R^2$={r2:.2f}", fontsize=32)
|
| 964 |
+
# Adjusting font sizes and setting font properties
|
| 965 |
+
plt.xlabel(f'AlphaFold-pLDDT',size=32)
|
| 966 |
+
plt.ylabel(f'FusOn-pLM-Diso',size=32)
|
| 967 |
+
plt.title(f"% Disordered (n={len(actual_values)})",size=32)
|
| 968 |
+
plt.xticks(fontsize=24)
|
| 969 |
+
plt.yticks(fontsize=24)
|
| 970 |
+
#plt.xlabel("Values")
|
| 971 |
+
#plt.ylabel("Density")
|
| 972 |
+
#plt.title(f"Density Plot of Actual vs Predicted Values (R^2 = {r2:.2f})")
|
| 973 |
+
plt.legend(prop={'size': 16})
|
| 974 |
+
plt.tight_layout()
|
| 975 |
+
plt.show()
|
| 976 |
+
plt.savefig(save_path, dpi=300)
|
| 977 |
+
|
| 978 |
+
def main():
|
| 979 |
+
set_font()
|
| 980 |
+
#output_dir = "results/test"
|
| 981 |
+
output_dir = "results/final"
|
| 982 |
+
seq_label_dict = pd.read_csv('splits/test_df.csv')
|
| 983 |
+
seq_ids_dict = dict(zip(seq_label_dict['Sequence'],seq_label_dict['IDs']))
|
| 984 |
+
seq_label_dict = dict(zip(seq_label_dict['Sequence'],seq_label_dict['Label']))
|
| 985 |
+
best_caid_model_results = pd.read_csv(f"{output_dir}/best_caid_model_results.csv")
|
| 986 |
+
make_auroc_curve(results_dir=output_dir,
|
| 987 |
+
seq_label_dict=seq_label_dict,
|
| 988 |
+
seq_ids_dict=seq_ids_dict,
|
| 989 |
+
path_to_results_of_interest="trained_models/fuson_plm/best/caid_hyperparam_screen_test_probs.csv",
|
| 990 |
+
model_alias="FusOn-pLM",
|
| 991 |
+
path_to_esm_results="trained_models/esm2_t33_650M_UR50D/best/caid_hyperparam_screen_test_probs.csv",
|
| 992 |
+
with_rankings=True)
|
| 993 |
+
|
| 994 |
+
caid2_test_data = pd.read_csv(f"splits/splits.csv")
|
| 995 |
+
caid2_test_data = caid2_test_data.loc[caid2_test_data['Split']=='Test']
|
| 996 |
+
caid2_test_labels = caid2_test_data['Label'].tolist()
|
| 997 |
+
caid2_test_ids = caid2_test_data['IDs'].tolist()
|
| 998 |
+
# fusions, heads, and tails
|
| 999 |
+
fusion_ht_data = prep_data_for_ht_disorder_comparison()
|
| 1000 |
+
os.makedirs("processed_data/figures",exist_ok=True)
|
| 1001 |
+
|
| 1002 |
+
head_data = fusion_ht_data.drop_duplicates(['hg_seq']).reset_index(drop=True)
|
| 1003 |
+
head_labels = head_data['hg_label'].tolist()
|
| 1004 |
+
head_ids = head_data['hgUniProt'].tolist()
|
| 1005 |
+
tail_data = fusion_ht_data.drop_duplicates(['tg_seq']).reset_index(drop=True)
|
| 1006 |
+
tail_labels = tail_data['tg_label'].tolist()
|
| 1007 |
+
tail_ids = tail_data['tgUniProt'].tolist()
|
| 1008 |
+
fusion_data = fusion_ht_data.drop_duplicates(['Fusion_Seq']).reset_index(drop=True)
|
| 1009 |
+
fusion_labels = fusion_data['fusion_label'].tolist()
|
| 1010 |
+
fusion_ids = fusion_data['seq_id'].tolist()
|
| 1011 |
+
|
| 1012 |
+
plt.rc('text', usetex=False)
|
| 1013 |
+
math_part = r"$n$"
|
| 1014 |
+
|
| 1015 |
+
os.makedirs("processed_data/figures/histograms",exist_ok=True)
|
| 1016 |
+
plot_disorder_content_hist(caid2_test_labels, caid2_test_ids, title=f"CAID2 Disorder-NOX ({math_part}={len(caid2_test_labels):,})", color="black", savepath='processed_data/figures/histograms/disorder_nox_histogram.png')
|
| 1017 |
+
plot_disorder_content_hist(head_labels, head_ids, title=f"Head Proteins ({math_part}={len(head_labels):,})", color="#df8385", savepath='processed_data/figures/histograms/heads_histogram.png')
|
| 1018 |
+
plot_disorder_content_hist(tail_labels, tail_ids, title=f"Tail Proteins ({math_part}={len(tail_labels):,})", color="#6ea4da", savepath='processed_data/figures/histograms/tails_histogram.png')
|
| 1019 |
+
plot_disorder_content_hist(fusion_labels, fusion_ids, title=f"Fusion Oncoproteins ({math_part}={len(fusion_labels):,})", color="mediumpurple", savepath='processed_data/figures/histograms/fusions_histogram.png')
|
| 1020 |
+
|
| 1021 |
+
os.makedirs("processed_data/figures/fusion_disorder",exist_ok=True)
|
| 1022 |
+
make_all_favorite_fusion_pLDDT_plots([
|
| 1023 |
+
"EWSR1::FLI1",
|
| 1024 |
+
"PAX3::FOXO1",
|
| 1025 |
+
"EML4::ALK",
|
| 1026 |
+
"SS18::SSX1"],
|
| 1027 |
+
left_to_right=True)
|
| 1028 |
+
|
| 1029 |
+
if __name__ == "__main__":
|
| 1030 |
+
main()
|
fuson_plm/benchmarking/caid/process_fusion_structures.py
ADDED
|
@@ -0,0 +1,799 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
# Process fusion structures and the structures of their head and tail proteins for pLDDTs
|
| 2 |
+
|
| 3 |
+
import requests
|
| 4 |
+
import json
|
| 5 |
+
import pandas as pd
|
| 6 |
+
import numpy as np
|
| 7 |
+
|
| 8 |
+
import requests
|
| 9 |
+
import re
|
| 10 |
+
import os
|
| 11 |
+
import shutil
|
| 12 |
+
|
| 13 |
+
from Bio.PDB import MMCIFParser
|
| 14 |
+
import Bio.PDB as PDB
|
| 15 |
+
from Bio import pairwise2
|
| 16 |
+
from Bio.pairwise2 import format_alignment
|
| 17 |
+
from bs4 import BeautifulSoup
|
| 18 |
+
import pdb
|
| 19 |
+
|
| 20 |
+
from fuson_plm.utils.logging import log_update, open_logfile
|
| 21 |
+
|
| 22 |
+
#@markdown Define AlphaFoldStructure class
|
| 23 |
+
class AlphaFoldStructure:
|
| 24 |
+
'''
|
| 25 |
+
This class processes an mmCIF file, either uploaded or downloaded from the AlphaFold2 database, to provide comprehensive information.
|
| 26 |
+
'''
|
| 27 |
+
def __init__(self, fold_path=None, uniprot_to_download=None, uniprot_output_dir= None, secondary_structure_types=None):
|
| 28 |
+
# If the user provided a PDB path, convert their file to mmcif. Isolate the suffix
|
| 29 |
+
if fold_path is not None:
|
| 30 |
+
fold_fname = fold_path.split('/')[-1]
|
| 31 |
+
prefix, suffix = fold_fname.split('.')
|
| 32 |
+
|
| 33 |
+
if suffix == 'pdb': # convert to cif
|
| 34 |
+
# make a directory for converted cif files
|
| 35 |
+
conversion_path = 'mmcif_converted_files'
|
| 36 |
+
if not(os.path.exists(conversion_path)):
|
| 37 |
+
os.makedirs(conversion_path)
|
| 38 |
+
|
| 39 |
+
fold_path = self.__convert_pdb_to_mmcif__(fold_path, f'{conversion_path}/{prefix}.cif')
|
| 40 |
+
|
| 41 |
+
self.file_path = fold_path
|
| 42 |
+
|
| 43 |
+
# If user provided a uniprot ID to download, download it and save it as the file path so it can be processed
|
| 44 |
+
if uniprot_to_download is not None:
|
| 45 |
+
if fold_path is not None:
|
| 46 |
+
log_update("WARNING: both a fold_path and a uniprot_to_download were provided. Running default: downloading the CIF file for provided UniProt ID.")
|
| 47 |
+
self.file_path = self.__download_mmCIF(uniprot_to_download, output_path=uniprot_output_dir)
|
| 48 |
+
|
| 49 |
+
# Either they provide acceptable secondary structure types, or query the internet for them
|
| 50 |
+
if secondary_structure_types is None:
|
| 51 |
+
self.secondary_structure_types = self.__pull_secondary_structure_types()
|
| 52 |
+
else:
|
| 53 |
+
self.secondary_structure_types = secondary_structure_types
|
| 54 |
+
|
| 55 |
+
# If there's a CIF file, initialize the object
|
| 56 |
+
if self.file_path:
|
| 57 |
+
self.cif_lines = self.__parse_cif()
|
| 58 |
+
self.secondary_structures = self.__extract_secondary_structures()
|
| 59 |
+
self.structure_dict = self.__calc_pLDDTs()
|
| 60 |
+
self.sequence = self.structure_dict['seq']
|
| 61 |
+
self.plddts = self.structure_dict['res_pLDDTs']
|
| 62 |
+
self.avg_pLDDT = self.structure_dict['avg_pLDDT']
|
| 63 |
+
self.residues_df = self.__create_residues_summary_dataframe()
|
| 64 |
+
self.secondary_structures_df = self.__create_secondary_structures_summary_dataframe()
|
| 65 |
+
# Otherwise, print an error.
|
| 66 |
+
else:
|
| 67 |
+
log_update("ERROR: structure could not be created. No CIF file found.")
|
| 68 |
+
|
| 69 |
+
def __convert_pdb_to_mmcif__(self, pdb_filename, mmcif_filename):
|
| 70 |
+
parser = PDB.PDBParser()
|
| 71 |
+
structure = parser.get_structure('structure', pdb_filename)
|
| 72 |
+
|
| 73 |
+
io = PDB.MMCIFIO()
|
| 74 |
+
io.set_structure(structure)
|
| 75 |
+
io.save(mmcif_filename)
|
| 76 |
+
return mmcif_filename
|
| 77 |
+
|
| 78 |
+
def __download_mmCIF(self, uniprot_id, output_path=None):
|
| 79 |
+
'''
|
| 80 |
+
Download mmCIF file with provided uniprot_id and optional output_path for the downloaded file.
|
| 81 |
+
|
| 82 |
+
Return: path to downloaded file if successful, None otherwise
|
| 83 |
+
'''
|
| 84 |
+
full_file_name = f"AF-{uniprot_id}-F1-model_v4.cif" # define file name that will be found on the AlphaFold2 database.
|
| 85 |
+
# if output path not provided, just save locally under full_file_name
|
| 86 |
+
if output_path is None:
|
| 87 |
+
output_path = full_file_name
|
| 88 |
+
else:
|
| 89 |
+
output_path = f"{output_path}/{full_file_name}"
|
| 90 |
+
|
| 91 |
+
# request the URL for the file
|
| 92 |
+
url = f"https://alphafold.ebi.ac.uk/files/{full_file_name}"
|
| 93 |
+
response = requests.get(url)
|
| 94 |
+
|
| 95 |
+
if response.status_code == 200:
|
| 96 |
+
with open(output_path, 'wb') as file:
|
| 97 |
+
file.write(response.content)
|
| 98 |
+
#log_update(f"File downloaded successfully and saved as {output_path}")
|
| 99 |
+
else:
|
| 100 |
+
log_update(f"Failed to download file. Status code: {response.status_code}")
|
| 101 |
+
return None
|
| 102 |
+
|
| 103 |
+
return output_path
|
| 104 |
+
|
| 105 |
+
def __pull_secondary_structure_types(self):
|
| 106 |
+
'''
|
| 107 |
+
Pull a dictionary of secondary structure types and their descriptions from the PDB mmCIF website (necessary for annotating the CIF file)
|
| 108 |
+
Only called if the user does not provide such a dictionary themselves.
|
| 109 |
+
'''
|
| 110 |
+
|
| 111 |
+
# request the .html tree from the website with all secondary structure terms
|
| 112 |
+
url = "https://mmcif.wwpdb.org/dictionaries/mmcif_pdbx_v50.dic/Items/_struct_conf_type.id.html"
|
| 113 |
+
response = requests.get(url)
|
| 114 |
+
|
| 115 |
+
if response.status_code != 200:
|
| 116 |
+
raise Exception("Failed to retrieve mmCIF dictionary")
|
| 117 |
+
|
| 118 |
+
# Parse the response content
|
| 119 |
+
soup = BeautifulSoup(response.content, 'html.parser')
|
| 120 |
+
|
| 121 |
+
# Debug: Print the soup to understand the structure
|
| 122 |
+
# log_update(soup.prettify())
|
| 123 |
+
# write the prettified soup to a txt file
|
| 124 |
+
#with open('mmcif_dictionary.txt', 'w') as f:
|
| 125 |
+
# f.write(soup.prettify())
|
| 126 |
+
|
| 127 |
+
# Find the h4 header with the class "panel-title" and text "Controlled Vocabulary"
|
| 128 |
+
header = soup.find('h4', class_='panel-title')
|
| 129 |
+
if header is None or 'Controlled Vocabulary' not in header.text:
|
| 130 |
+
raise Exception("Could not find the 'Controlled Vocabulary' header")
|
| 131 |
+
|
| 132 |
+
# Debug: Print the found header
|
| 133 |
+
#log_update(f"Found header: {header}")
|
| 134 |
+
|
| 135 |
+
# The table should be the next sibling of the header
|
| 136 |
+
table = header.find_next('table')
|
| 137 |
+
if table is None:
|
| 138 |
+
raise Exception("Could not find the table following the 'Controlled Vocabulary' header")
|
| 139 |
+
|
| 140 |
+
# Debug: Print the found table (only the opening <table> tag)
|
| 141 |
+
#log_update(f"Found table (showing header line): {str(table).split('<thead')[0]}")
|
| 142 |
+
|
| 143 |
+
# Iterate through rows in the table and process each entry
|
| 144 |
+
secondary_structure_types = {}
|
| 145 |
+
rows = table.find_all('tr')
|
| 146 |
+
for row in rows[1:]: # Skip the header row
|
| 147 |
+
cols = row.find_all('td')
|
| 148 |
+
if len(cols) > 1:
|
| 149 |
+
type_id = cols[0].text.strip()
|
| 150 |
+
description = cols[1].text.replace('\t', ' ').strip()
|
| 151 |
+
|
| 152 |
+
# Replace multiple spaces with a single space
|
| 153 |
+
description = re.sub(' +', ' ', description)
|
| 154 |
+
|
| 155 |
+
# If this is a protein secondary structure (the table also contains nucleic acid structures), add it to teh dictionary
|
| 156 |
+
if '(protein)' in description:
|
| 157 |
+
secondary_structure_types[type_id] = description
|
| 158 |
+
|
| 159 |
+
return secondary_structure_types
|
| 160 |
+
|
| 161 |
+
def get_secondary_structure_types(self):
|
| 162 |
+
'''
|
| 163 |
+
Display secondary structure types
|
| 164 |
+
'''
|
| 165 |
+
log_update("Secondary Structure Types in mmCIF files:")
|
| 166 |
+
for ss_type, description in self.secondary_structure_types.items():
|
| 167 |
+
log_update(f"{ss_type}: {description}")
|
| 168 |
+
|
| 169 |
+
return self.secondary_structure_types
|
| 170 |
+
|
| 171 |
+
def __parse_cif(self):
|
| 172 |
+
'''
|
| 173 |
+
Read cif file lines from self.file_path
|
| 174 |
+
'''
|
| 175 |
+
with open(self.file_path, 'r') as file:
|
| 176 |
+
lines = file.readlines()
|
| 177 |
+
return lines
|
| 178 |
+
|
| 179 |
+
def __extract_secondary_structures(self):
|
| 180 |
+
'''
|
| 181 |
+
Iterate through the lines of the cif files to find each secondary structure.
|
| 182 |
+
Returns a tuple for each amino acid that has a secondary structure annotation. Tuple contains:
|
| 183 |
+
1. Structure Type (e.g. STRN)
|
| 184 |
+
2. Structure ID (e.g. STRN1)
|
| 185 |
+
3. Description (e.g. beta strand)
|
| 186 |
+
4. Position (e.g. 3)
|
| 187 |
+
'''
|
| 188 |
+
secondary_structures = []
|
| 189 |
+
parsing_secondary_structure = False
|
| 190 |
+
|
| 191 |
+
# iterate throhugh cif lines
|
| 192 |
+
for line in self.cif_lines:
|
| 193 |
+
# hone in on the right section of the cif file
|
| 194 |
+
if line.startswith("_struct_conf.conf_type_id"):
|
| 195 |
+
parsing_secondary_structure = True
|
| 196 |
+
continue
|
| 197 |
+
# if we're in the right section...
|
| 198 |
+
if parsing_secondary_structure:
|
| 199 |
+
if line.startswith("#"):
|
| 200 |
+
parsing_secondary_structure = False # no longer in the right section
|
| 201 |
+
continue
|
| 202 |
+
# still in the right section
|
| 203 |
+
columns = line.split()
|
| 204 |
+
# iterate through columns to find each piece of info we need
|
| 205 |
+
if len(columns) >= 7:
|
| 206 |
+
sec_struc_type = columns[6]
|
| 207 |
+
sec_struc_id = columns[13]
|
| 208 |
+
start_res = int(columns[2])
|
| 209 |
+
end_res = int(columns[9])
|
| 210 |
+
sec_struc_name = self.secondary_structure_types.get(sec_struc_type, 'Unknown')
|
| 211 |
+
# make tuple for this position in the sequence
|
| 212 |
+
for pos in range(start_res, end_res + 1):
|
| 213 |
+
secondary_structures.append((sec_struc_type, sec_struc_id, sec_struc_name, pos))
|
| 214 |
+
|
| 215 |
+
return secondary_structures
|
| 216 |
+
|
| 217 |
+
def __calc_pLDDTs(self):
|
| 218 |
+
'''
|
| 219 |
+
This method iterates through the cif file to return a dictionary with a few key pieces of info:
|
| 220 |
+
1. Sequence
|
| 221 |
+
2. pLDDTs for each residue
|
| 222 |
+
3. Average pLDDT
|
| 223 |
+
'''
|
| 224 |
+
|
| 225 |
+
# define dictionary needed to translate into single-letter AA code
|
| 226 |
+
aa_dict = {
|
| 227 |
+
"ALA": "A", "CYS": "C", "ASP": "D", "GLU": "E", "PHE": "F",
|
| 228 |
+
"GLY": "G", "HIS": "H", "ILE": "I", "LYS": "K", "LEU": "L",
|
| 229 |
+
"MET": "M", "ASN": "N", "PRO": "P", "GLN": "Q", "ARG": "R",
|
| 230 |
+
"SER": "S", "THR": "T", "VAL": "V", "TRP": "W", "TYR": "Y"
|
| 231 |
+
}
|
| 232 |
+
|
| 233 |
+
parser = MMCIFParser(QUIET=True) # create a parser
|
| 234 |
+
data = parser.get_structure("structure", self.file_path) # parse structure
|
| 235 |
+
|
| 236 |
+
# count models and chains (should be 1 model and 1 chain; don't use this class to parse a complex)
|
| 237 |
+
model = data.get_models()
|
| 238 |
+
models = list(model)
|
| 239 |
+
chains = list(models[0].get_chains())
|
| 240 |
+
|
| 241 |
+
# iterate through the chains and get amino acid letters and pLDDTs
|
| 242 |
+
all_pLDDTs = []
|
| 243 |
+
for n in range(len(chains)):
|
| 244 |
+
chainname = chr(n + 65) # turn chain number into letter (e.g. 1 --> "A" so we have Chain A instead of Chain 1)
|
| 245 |
+
residues = list(chains[n].get_residues()) # extract all residues
|
| 246 |
+
seq = ''
|
| 247 |
+
pLDDTs = [0] * len(residues) # initialize empty pLDDT array for this chain
|
| 248 |
+
|
| 249 |
+
# iterate through all residues in this chain
|
| 250 |
+
for i in range(len(residues)):
|
| 251 |
+
r = residues[i]
|
| 252 |
+
# which amino acid is here?
|
| 253 |
+
try:
|
| 254 |
+
seq += aa_dict[r.get_resname()]
|
| 255 |
+
# error if it's not a real amino acid
|
| 256 |
+
except KeyError:
|
| 257 |
+
log_update('residue name invalid')
|
| 258 |
+
break
|
| 259 |
+
|
| 260 |
+
# look at each atom. Get its pLDDT (bfactor). make sure bfactor for all atoms within one residue are equal.
|
| 261 |
+
atoms = list(r.get_atoms())
|
| 262 |
+
bfactor = atoms[0].get_bfactor()
|
| 263 |
+
for a in range(len(atoms)):
|
| 264 |
+
# if not all atoms within an AA have the same pLDDT, error.
|
| 265 |
+
if atoms[a].get_bfactor() != bfactor:
|
| 266 |
+
break
|
| 267 |
+
|
| 268 |
+
pLDDTs[i] = bfactor # add pLDDT for this residue to the list.
|
| 269 |
+
|
| 270 |
+
all_pLDDTs.extend(pLDDTs) # add pLDDTs for this chain to list of all pLDDTs
|
| 271 |
+
|
| 272 |
+
avg_pLDDT = np.mean(all_pLDDTs) # average pLDDTs across all chains
|
| 273 |
+
return_dict = {
|
| 274 |
+
'avg_pLDDT': round(avg_pLDDT, 2),
|
| 275 |
+
'res_pLDDTs': all_pLDDTs,
|
| 276 |
+
'seq': seq
|
| 277 |
+
}
|
| 278 |
+
return return_dict
|
| 279 |
+
|
| 280 |
+
def __create_residues_summary_dataframe(self):
|
| 281 |
+
'''
|
| 282 |
+
Create a dataframe that summarizes the secondary structure information for each residue.
|
| 283 |
+
Columns:
|
| 284 |
+
1. Position: amino acid position (e.g. 3)
|
| 285 |
+
2. Residue: amino acid 1-letter code (e.g. A)
|
| 286 |
+
3. pLDDT: alphafold2's pLDDT score for this residue to 2 decimal places (e.g. 77.54)
|
| 287 |
+
4. Structure Type: type of secondary structure (e.g. STRN)
|
| 288 |
+
5. Structure ID: ID of this secondary structure (e.g. STRN1)
|
| 289 |
+
5. Description: description of this secondary structure (e.g. beta strand)
|
| 290 |
+
6. Disordered: is this residue disordered or not? A residue is not disordered if it's in a HELX or STRN. (True/False)
|
| 291 |
+
|
| 292 |
+
'''
|
| 293 |
+
# Convert the secondary structures to a dataframe
|
| 294 |
+
df_secondary_structures = pd.DataFrame(self.secondary_structures, columns=['Structure Type', 'Structure ID', 'Description', 'Position'])
|
| 295 |
+
|
| 296 |
+
# Add Residue and pLDDT columns to the dataframe
|
| 297 |
+
df_temp = pd.DataFrame(
|
| 298 |
+
data={
|
| 299 |
+
'Position': list(range(1, len(self.sequence) + 1)),
|
| 300 |
+
'Residue': list(self.sequence),
|
| 301 |
+
'pLDDT': self.plddts
|
| 302 |
+
})
|
| 303 |
+
|
| 304 |
+
df_secondary_structures = pd.merge(df_secondary_structures, df_temp, on='Position', how='right')
|
| 305 |
+
# Determine if each residue is disordered or not based on what Structure Type it's in. If helix or strand, it's ordered. If anything else or NaN, it's disordered.
|
| 306 |
+
df_secondary_structures['Disordered'] = df_secondary_structures['Structure Type'].apply(
|
| 307 |
+
lambda x: False if (type(x)==str and (('HELX' in x) or ('STRN' in x))) else True
|
| 308 |
+
)
|
| 309 |
+
|
| 310 |
+
return df_secondary_structures
|
| 311 |
+
|
| 312 |
+
def __create_secondary_structures_summary_dataframe(self):
|
| 313 |
+
'''
|
| 314 |
+
Create a dataframe grouped by each Structure ID, providing a summary of each secondary structure in the chain.
|
| 315 |
+
Columns:
|
| 316 |
+
1. Structure ID: ID of this secondary structure (e.g. STRN1)
|
| 317 |
+
2. Start: start position of this secondary structure (e.g. 3)
|
| 318 |
+
3. End: end position of this secondary structure (e.g. 12)
|
| 319 |
+
4. Start Residue: amino acid 1-letter code of the start position (e.g. A)
|
| 320 |
+
5. End Residue: amino acid 1-letter code of the end position (e.g. L)
|
| 321 |
+
6. Disordered: is this residue disordered or not? A residue is not disordered if it's in a HELX or STRN. (True/False)
|
| 322 |
+
7. Description: description of this secondary structure (e.g. beta strand)
|
| 323 |
+
8. Structure Type: type of secondary structure (e.g. STRN)
|
| 324 |
+
9. avg_pLDDT: average pLDDT for this secondary structure (e.g. 77.54)
|
| 325 |
+
'''
|
| 326 |
+
|
| 327 |
+
# Apply groupby on self.residues_df to reorganize it by Structure ID
|
| 328 |
+
secondary_structures_df = self.residues_df.groupby('Structure ID').agg({
|
| 329 |
+
'Position': ['first', 'last'],
|
| 330 |
+
'Residue': ['first','last'],
|
| 331 |
+
'Disordered': 'first',
|
| 332 |
+
'Description': 'first',
|
| 333 |
+
'Structure Type': 'first',
|
| 334 |
+
'pLDDT': 'mean'
|
| 335 |
+
}).reset_index()
|
| 336 |
+
|
| 337 |
+
# Flatten the multi-level columns
|
| 338 |
+
secondary_structures_df.columns = ['Structure ID', 'Start', 'End', 'Start Residue', 'End Residue', 'Disordered', 'Description', 'Structure Type', 'avg_pLDDT']
|
| 339 |
+
secondary_structures_df['avg_pLDDT'] = secondary_structures_df['avg_pLDDT'].round(2)
|
| 340 |
+
|
| 341 |
+
# Display the summarized DataFrame
|
| 342 |
+
return secondary_structures_df
|
| 343 |
+
|
| 344 |
+
def get_residues_df(self):
|
| 345 |
+
return self.residues_df
|
| 346 |
+
|
| 347 |
+
def get_secondary_structures_df(self):
|
| 348 |
+
return self.secondary_structures_df
|
| 349 |
+
|
| 350 |
+
def get_full_sequence(self):
|
| 351 |
+
return ''.join([res for res in self.residues_df['Residue']])
|
| 352 |
+
|
| 353 |
+
def get_average_plddt(self):
|
| 354 |
+
plddt_values = [plddt for plddt in self.residues_df['pLDDT'] if plddt is not None]
|
| 355 |
+
return sum(plddt_values) / len(plddt_values) if plddt_values else None
|
| 356 |
+
|
| 357 |
+
def pull_secondary_structure_types():
|
| 358 |
+
url = "https://mmcif.wwpdb.org/dictionaries/mmcif_pdbx_v50.dic/Items/_struct_conf_type.id.html"
|
| 359 |
+
response = requests.get(url)
|
| 360 |
+
|
| 361 |
+
if response.status_code != 200:
|
| 362 |
+
raise Exception("Failed to retrieve mmCIF dictionary")
|
| 363 |
+
|
| 364 |
+
soup = BeautifulSoup(response.content, 'html.parser')
|
| 365 |
+
|
| 366 |
+
# Debug: Print the soup to understand the structure
|
| 367 |
+
# log_update(soup.prettify())
|
| 368 |
+
# write the prettified soup to a txt file
|
| 369 |
+
with open('mmcif_dictionary.txt', 'w') as f:
|
| 370 |
+
f.write(soup.prettify())
|
| 371 |
+
|
| 372 |
+
# Find the h4 header with the class "panel-title" and text "Controlled Vocabulary"
|
| 373 |
+
header = soup.find('h4', class_='panel-title')
|
| 374 |
+
if header is None or 'Controlled Vocabulary' not in header.text:
|
| 375 |
+
raise Exception("Could not find the 'Controlled Vocabulary' header")
|
| 376 |
+
|
| 377 |
+
# Debug: Print the found header
|
| 378 |
+
#log_update(f"Found header: {header}")
|
| 379 |
+
|
| 380 |
+
# The table should be the next sibling of the header
|
| 381 |
+
table = header.find_next('table')
|
| 382 |
+
if table is None:
|
| 383 |
+
raise Exception("Could not find the table following the 'Controlled Vocabulary' header")
|
| 384 |
+
|
| 385 |
+
# Debug: Print the found table (only the opening <table> tag)
|
| 386 |
+
#log_update(f"Found table (showing header line): {str(table).split('<thead')[0]}")
|
| 387 |
+
|
| 388 |
+
secondary_structure_types = {}
|
| 389 |
+
rows = table.find_all('tr')
|
| 390 |
+
for row in rows[1:]: # Skip the header row
|
| 391 |
+
cols = row.find_all('td')
|
| 392 |
+
if len(cols) > 1:
|
| 393 |
+
type_id = cols[0].text.strip()
|
| 394 |
+
description = cols[1].text.replace('\t', ' ').strip()
|
| 395 |
+
|
| 396 |
+
# Replace multiple spaces with a single space
|
| 397 |
+
description = re.sub(' +', ' ', description)
|
| 398 |
+
|
| 399 |
+
if '(protein)' in description:
|
| 400 |
+
secondary_structure_types[type_id] = description
|
| 401 |
+
|
| 402 |
+
return secondary_structure_types
|
| 403 |
+
|
| 404 |
+
# Process structures downloaded from FusionPDB
|
| 405 |
+
def process_fusionpdb_fusion_files(files, level_2_3_structure_info, folder, save_path=None):
|
| 406 |
+
# get secondary structure types so we can process PDBs
|
| 407 |
+
secondary_structure_types = pull_secondary_structure_types()
|
| 408 |
+
|
| 409 |
+
# Initialize 3 columns to store structural info - the AA seq in the fold (should match), the Avg pLDDT, and the per-residue pLDDTs (comma-separated, 2 decimal pts.)
|
| 410 |
+
level_2_3_structure_info['Fold AA seq'] = ['']*len(level_2_3_structure_info)
|
| 411 |
+
level_2_3_structure_info['Avg pLDDT'] = [0]*len(level_2_3_structure_info)
|
| 412 |
+
level_2_3_structure_info['pLDDTs'] = ['']*len(level_2_3_structure_info)
|
| 413 |
+
|
| 414 |
+
# pre-loop processed
|
| 415 |
+
pre_loop_processed = []
|
| 416 |
+
if os.path.exists(save_path):
|
| 417 |
+
pre_loop_processed = pd.read_csv(save_path)
|
| 418 |
+
pre_loop_processed = pre_loop_processed['Structure Link'].tolist()
|
| 419 |
+
pre_loop_processed = [x.split('/')[-1] for x in pre_loop_processed]
|
| 420 |
+
log_update(f"Total structures already processed: {len(pre_loop_processed)}")
|
| 421 |
+
|
| 422 |
+
log_update("\nProcessing fusion structures...")
|
| 423 |
+
# only process structures we haven't processed yet
|
| 424 |
+
for i, structure in enumerate(files):
|
| 425 |
+
log_update(f'\tProcessing #{i+1}: {structure}')
|
| 426 |
+
|
| 427 |
+
# make sure we haven't already processed it and aren't wasting time
|
| 428 |
+
if structure in pre_loop_processed:
|
| 429 |
+
log_update(f"\t\tAlready processed. Continuing...")
|
| 430 |
+
continue
|
| 431 |
+
|
| 432 |
+
# create AlphaFoldStructure object
|
| 433 |
+
obj = AlphaFoldStructure(fold_path=f'{folder}/{structure}', secondary_structure_types=secondary_structure_types)
|
| 434 |
+
aa_seq = obj.get_full_sequence()
|
| 435 |
+
avg_plddt = obj.get_average_plddt()
|
| 436 |
+
residues_df = obj.get_residues_df()
|
| 437 |
+
all_plddts = ",".join(residues_df['pLDDT'].astype(str).tolist())
|
| 438 |
+
|
| 439 |
+
log_update(f"\t\tAvg pLDDT: {round(avg_plddt,2)}\tFold AA seq: {aa_seq}\tFirst 5 pLDDTs: {','.join(all_plddts.split(',')[0:5])}")
|
| 440 |
+
|
| 441 |
+
level_2_3_structure_info.loc[level_2_3_structure_info['Structure Link'].str.contains(f"/{structure}"), 'Fold AA seq'] = aa_seq
|
| 442 |
+
level_2_3_structure_info.loc[level_2_3_structure_info['Structure Link'].str.contains(f"/{structure}"), 'Avg pLDDT'] = avg_plddt
|
| 443 |
+
level_2_3_structure_info.loc[level_2_3_structure_info['Structure Link'].str.contains(f"/{structure}"), 'pLDDTs'] = all_plddts
|
| 444 |
+
|
| 445 |
+
# write level_2_3_structure_info to csv
|
| 446 |
+
cur_df = level_2_3_structure_info.loc[level_2_3_structure_info['Structure Link'].str.contains(f"/{structure}")].reset_index(drop=True)
|
| 447 |
+
if os.path.exists(save_path):
|
| 448 |
+
cur_df.to_csv(save_path,mode='a',header=False,index=False)
|
| 449 |
+
else:
|
| 450 |
+
cur_df.to_csv(save_path,index=False)
|
| 451 |
+
|
| 452 |
+
# now reload the completed dataframe
|
| 453 |
+
level_2_3_structure_info = pd.read_csv(save_path)
|
| 454 |
+
return level_2_3_structure_info
|
| 455 |
+
|
| 456 |
+
def process_fusionpdb_head_tail_files(ht, save_path='heads_and_tails_structures_processed.csv'):
|
| 457 |
+
# ht is a list of head and tail proteins we have to process.
|
| 458 |
+
log_update("\nProcessing head and tail structures...")
|
| 459 |
+
|
| 460 |
+
# get secondary structure types so we can process PDBs
|
| 461 |
+
secondary_structure_types = pull_secondary_structure_types()
|
| 462 |
+
|
| 463 |
+
# make directory to save alphafold DB structures of heads and tails
|
| 464 |
+
os.makedirs('raw_data/fusionpdb/head_tail_af2db_structures',exist_ok=True)
|
| 465 |
+
|
| 466 |
+
# pre-loop processed
|
| 467 |
+
pre_loop_processed = []
|
| 468 |
+
if os.path.exists(save_path):
|
| 469 |
+
pre_loop_processed = pd.read_csv(save_path)
|
| 470 |
+
pre_loop_processed = pre_loop_processed['UniProtID'].tolist()
|
| 471 |
+
log_update(f"Heads and tails already processed: {len(pre_loop_processed)}")
|
| 472 |
+
|
| 473 |
+
ht_structures_df = pd.DataFrame(
|
| 474 |
+
data = {
|
| 475 |
+
'UniProtID': ['']*len(ht),
|
| 476 |
+
'Avg pLDDT': ['']*len(ht),
|
| 477 |
+
'All pLDDTs': ['']*len(ht),
|
| 478 |
+
'Seq': ['']*len(ht)
|
| 479 |
+
}
|
| 480 |
+
)
|
| 481 |
+
|
| 482 |
+
for i, uniprotid in enumerate(ht):
|
| 483 |
+
log_update(f'\tProcessing #{i+1}: {uniprotid}')
|
| 484 |
+
aa_seq, avg_plddt, all_plddts = None, None, None
|
| 485 |
+
|
| 486 |
+
# make sure we haven't processed it yet!
|
| 487 |
+
if uniprotid in pre_loop_processed:
|
| 488 |
+
log_update(f"\t\tAlready processed. Continuing")
|
| 489 |
+
continue
|
| 490 |
+
|
| 491 |
+
try:
|
| 492 |
+
obj = AlphaFoldStructure(uniprot_to_download=uniprotid, secondary_structure_types=secondary_structure_types,
|
| 493 |
+
uniprot_output_dir='raw_data/fusionpdb/head_tail_af2db_structures')
|
| 494 |
+
aa_seq = obj.get_full_sequence()
|
| 495 |
+
avg_plddt = obj.get_average_plddt()
|
| 496 |
+
residues_df = obj.get_residues_df()
|
| 497 |
+
all_plddts = ",".join(residues_df['pLDDT'].astype(str).tolist())
|
| 498 |
+
|
| 499 |
+
log_update(f"\t\tAvg pLDDT: {round(avg_plddt,2)}\tFold AA seq: {aa_seq}\tFirst 5 pLDDTs: {','.join(all_plddts.split(',')[0:5])}")
|
| 500 |
+
|
| 501 |
+
except:
|
| 502 |
+
log_update(f"\t\tAvg pLDDT: {None}\tFold AA seq: {None}\tFirst 5 pLDDTs: {None}")
|
| 503 |
+
|
| 504 |
+
# Fill in info for combined ht df
|
| 505 |
+
ht_structures_df.loc[i, 'UniProtID'] = uniprotid
|
| 506 |
+
ht_structures_df.loc[i, 'Avg pLDDT'] = avg_plddt
|
| 507 |
+
ht_structures_df.loc[i, 'All pLDDTs'] = all_plddts
|
| 508 |
+
ht_structures_df.loc[i, 'Seq'] = aa_seq
|
| 509 |
+
|
| 510 |
+
# write level_2_3_structure_info to csv
|
| 511 |
+
cur_df = pd.DataFrame(ht_structures_df.iloc[i,:]).T.reset_index(drop=True)
|
| 512 |
+
if os.path.exists(save_path):
|
| 513 |
+
cur_df.to_csv(save_path,mode='a',header=False,index=False)
|
| 514 |
+
else:
|
| 515 |
+
cur_df.to_csv(save_path,index=False)
|
| 516 |
+
|
| 517 |
+
# ensure we got everything
|
| 518 |
+
ht_structures_df = pd.read_csv(save_path)
|
| 519 |
+
level_2_3 = pd.read_csv(f'processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_head_tail_info.csv')
|
| 520 |
+
level_2_3['FusionGene'] = level_2_3['FusionGene'].str.replace('-','::')
|
| 521 |
+
heads = level_2_3['HGUniProtAcc'].tolist()
|
| 522 |
+
tails = level_2_3['TGUniProtAcc'].tolist()
|
| 523 |
+
ht = heads + tails
|
| 524 |
+
ht = set([x for x in ht if type(x)==str])
|
| 525 |
+
ht = set(','.join(ht).split(','))
|
| 526 |
+
|
| 527 |
+
log_update(f"total heads and tails: {len(ht)}")
|
| 528 |
+
log_update(f"total processed: {len(ht_structures_df)}\t{len(ht_structures_df['UniProtID'].unique())}")
|
| 529 |
+
|
| 530 |
+
# which ones are missing?
|
| 531 |
+
missing = set(ht) - set(ht_structures_df['UniProtID'].unique())
|
| 532 |
+
log_update(f"missing: {len(missing)}")
|
| 533 |
+
log_update(missing)
|
| 534 |
+
|
| 535 |
+
# Some heads and tails are not in the alphxwafold database. I folded these myself, externally.
|
| 536 |
+
ht_structures_df = ht_structures_df.replace('',np.nan)
|
| 537 |
+
need_to_fold = ht_structures_df[ht_structures_df['Avg pLDDT'].isna()]['UniProtID'].tolist()
|
| 538 |
+
with open('processed_data/fusionpdb/intermediates/uniprotids_not_in_afdb.txt','w') as f:
|
| 539 |
+
for uniprotid in need_to_fold:
|
| 540 |
+
f.write(f'{uniprotid}\n')
|
| 541 |
+
|
| 542 |
+
idmap = pd.read_csv(f'raw_data/fusionpdb/not_in_afdb_idmap.txt',sep='\t')
|
| 543 |
+
idmap = idmap[idmap['Entry'].isin(need_to_fold)].reset_index(drop=True)
|
| 544 |
+
idmap = idmap[['Entry','Sequence']].rename(columns={
|
| 545 |
+
'Entry': 'ID'})
|
| 546 |
+
idmap['Length'] = idmap['Sequence'].apply(len)
|
| 547 |
+
|
| 548 |
+
log_update("Investigating heads and tails that were not in the AF2 database:")
|
| 549 |
+
log_update(f"\tMin length: {min(idmap['Length'])}")
|
| 550 |
+
log_update(f"\tMax length: {max(idmap['Length'])}")
|
| 551 |
+
idmap = idmap.sort_values(by='Length',ascending=True).reset_index(drop=True)
|
| 552 |
+
|
| 553 |
+
# Q9NNW7
|
| 554 |
+
id='Q9NNW7'
|
| 555 |
+
if id in idmap['ID'].tolist():
|
| 556 |
+
ht_structures_df.loc[
|
| 557 |
+
ht_structures_df['UniProtID']=='Q9NNW7', 'Avg pLDDT'
|
| 558 |
+
] = 91.68
|
| 559 |
+
ht_structures_df.loc[
|
| 560 |
+
ht_structures_df['UniProtID']=='Q9NNW7', 'Seq'
|
| 561 |
+
] = idmap.loc[
|
| 562 |
+
idmap['ID']=='Q9NNW7', 'Sequence'
|
| 563 |
+
].item()
|
| 564 |
+
|
| 565 |
+
## Q16881
|
| 566 |
+
id='Q16881'
|
| 567 |
+
if id in idmap['ID'].tolist():
|
| 568 |
+
ht_structures_df.loc[
|
| 569 |
+
ht_structures_df['UniProtID']==id, 'Avg pLDDT'
|
| 570 |
+
] = 89.55
|
| 571 |
+
ht_structures_df.loc[
|
| 572 |
+
ht_structures_df['UniProtID']==id, 'Seq'
|
| 573 |
+
] = idmap.loc[
|
| 574 |
+
idmap['ID']==id, 'Sequence'
|
| 575 |
+
].item()
|
| 576 |
+
|
| 577 |
+
# Q86V15
|
| 578 |
+
id='Q86V15'
|
| 579 |
+
if id in idmap['ID'].tolist():
|
| 580 |
+
ht_structures_df.loc[
|
| 581 |
+
ht_structures_df['UniProtID']==id, 'Avg pLDDT'
|
| 582 |
+
] = 48.14
|
| 583 |
+
ht_structures_df.loc[
|
| 584 |
+
ht_structures_df['UniProtID']==id, 'Seq'
|
| 585 |
+
] = idmap.loc[
|
| 586 |
+
idmap['ID']==id, 'Sequence'
|
| 587 |
+
].item()
|
| 588 |
+
|
| 589 |
+
return ht_structures_df
|
| 590 |
+
|
| 591 |
+
def process_fusions_and_hts():
|
| 592 |
+
# Process the structures of fusion proteins downloaded from FusionPDB
|
| 593 |
+
level_2_3_structure_info_og = pd.read_csv('processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_structure_links.csv')
|
| 594 |
+
|
| 595 |
+
# figure out which ones we have
|
| 596 |
+
folder = 'raw_data/fusionpdb/structures'
|
| 597 |
+
# get all the structure files in folder
|
| 598 |
+
files = os.listdir(folder)
|
| 599 |
+
log_update(f"total pdbs: {len(files)}")
|
| 600 |
+
log_update(f"examples: {files[:5]}")
|
| 601 |
+
|
| 602 |
+
os.makedirs('processed_data/fusionpdb', exist_ok=True)
|
| 603 |
+
|
| 604 |
+
# process the full fusion pdbs
|
| 605 |
+
level_2_3_structure_info = process_fusionpdb_fusion_files(files, level_2_3_structure_info_og, folder, save_path='processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_structures_processed.csv')
|
| 606 |
+
|
| 607 |
+
# process the head and tail pdbs
|
| 608 |
+
level_2_3 = pd.read_csv(f'processed_data/fusionpdb/intermediates/giant_level2-3_fusion_protein_head_tail_info.csv')
|
| 609 |
+
level_2_3['FusionGene'] = level_2_3['FusionGene'].str.replace('-','::')
|
| 610 |
+
# Get the heads and tails, see how many unique proteins we're working with
|
| 611 |
+
heads = level_2_3['HGUniProtAcc'].tolist()
|
| 612 |
+
tails = level_2_3['TGUniProtAcc'].tolist()
|
| 613 |
+
ht = heads + tails
|
| 614 |
+
ht = set([x for x in ht if type(x)==str])
|
| 615 |
+
ht = set(','.join(ht).split(','))
|
| 616 |
+
log_update(f"Unique heads/tails: {len(ht)}")
|
| 617 |
+
|
| 618 |
+
heads_tails_analyzed = process_fusionpdb_head_tail_files(list(ht), save_path='processed_data/fusionpdb/heads_tails_structural_data.csv')
|
| 619 |
+
|
| 620 |
+
# In the level_2_3 database, we only have the fusions with documented heads and tails.
|
| 621 |
+
level_2 = pd.read_csv(f'raw_data/fusionpdb/FusionPDB_level2_curated_09_05_2024.csv')
|
| 622 |
+
level_3 = pd.read_csv(f'raw_data/fusionpdb/FusionPDB_level3_curated_09_05_2024.csv')
|
| 623 |
+
joined_23 = pd.concat([level_2,level_3]).reset_index(drop=True)
|
| 624 |
+
joined_23['FusionGene'] = joined_23['FusionGene'].str.replace('-','::') # use new notation with head::tail
|
| 625 |
+
log_update(f"\nnumber of duplicated fusion gene rows: {len(joined_23[joined_23['FusionGene'].duplicated()])}")
|
| 626 |
+
# make the dictionary
|
| 627 |
+
fo_gid_dict = dict(zip(joined_23['FusionGene'],joined_23['FusionGID']))
|
| 628 |
+
log_update(len(fo_gid_dict))
|
| 629 |
+
|
| 630 |
+
# let's clean giant level 2 and level 3
|
| 631 |
+
# first, drop anyting where Fold AA seq is nan. there is no fold.
|
| 632 |
+
level_2_3_structure_info_clean = level_2_3_structure_info.replace('',np.nan) # make sure there are nans where there should be
|
| 633 |
+
level_2_3_structure_info_clean = level_2_3_structure_info_clean.dropna(subset=['Fold AA seq']).reset_index(drop=True)
|
| 634 |
+
log_update(f"length of processed structure file: {len(level_2_3_structure_info_clean)}")
|
| 635 |
+
level_2_3_structure_info_clean['pLDDT'] = level_2_3_structure_info_clean['Avg pLDDT'].round(2)
|
| 636 |
+
level_2_3_structure_info_clean = level_2_3_structure_info_clean.drop(columns=['Avg pLDDT'])
|
| 637 |
+
level_2_3_structure_info_clean['FusionGene'] = level_2_3_structure_info_clean['FusionGene'].str.replace('-','::')
|
| 638 |
+
level_2_3_structure_info_clean['FusionGID'] = level_2_3_structure_info_clean['FusionGene'].apply(lambda x: fo_gid_dict[x])
|
| 639 |
+
|
| 640 |
+
# now let's use the FusionPDB database we processed as ground truth for sequence, rather than the webpage
|
| 641 |
+
log_update("Using FusionPDB as ground truth for sequences...")
|
| 642 |
+
raw_download = pd.read_csv('../../data/raw_data/FusionPDB.txt',sep='\t',header=None)
|
| 643 |
+
raw_download['FusionGene'] = raw_download[7]+ '::' + raw_download[11]
|
| 644 |
+
raw_download = raw_download.rename(columns={18:'Raw Download AA Seq'})
|
| 645 |
+
log_update(f"FusionPDB raw download size: {len(raw_download)}")
|
| 646 |
+
|
| 647 |
+
level_2_3_structure_info_clean_ids = set(level_2_3_structure_info_clean['FusionGene'].tolist())
|
| 648 |
+
level_2_3_structure_info_clean_seqs = set(level_2_3_structure_info_clean['Fold AA seq'].tolist())
|
| 649 |
+
raw_download_ids = set(raw_download['FusionGene'].tolist())
|
| 650 |
+
raw_download_seqs = set(raw_download['Raw Download AA Seq'].tolist())
|
| 651 |
+
log_update(f"Number of overlapping gene IDs: {len(level_2_3_structure_info_clean_ids.intersection(raw_download_ids))}")
|
| 652 |
+
log_update(f"Number of overlapping sequences: {len(level_2_3_structure_info_clean_seqs.intersection(raw_download_seqs))}")
|
| 653 |
+
# attempt a merge on Raw Download AA Seq with both. ofthe ogs
|
| 654 |
+
# Merging with the AlphaFold sequence
|
| 655 |
+
test_merge_1 = pd.merge(
|
| 656 |
+
level_2_3_structure_info_clean.rename(columns={'Fold AA seq': 'Raw Download AA Seq'}),
|
| 657 |
+
raw_download,
|
| 658 |
+
on=['FusionGene','Raw Download AA Seq'],
|
| 659 |
+
how='inner'
|
| 660 |
+
)
|
| 661 |
+
test_merge_1 = test_merge_1.drop(columns=['AA seq'])
|
| 662 |
+
test_merge_1['Seq Source'] = ['AlphaFold,Raw Download']*len(test_merge_1)
|
| 663 |
+
log_update(f"Merge on AlphaFold AA Seq and raw Download AA Seq. len={len(test_merge_1)}")
|
| 664 |
+
# Merging with the webpage sequence
|
| 665 |
+
test_merge_2 = pd.merge(
|
| 666 |
+
level_2_3_structure_info_clean.rename(columns={'AA seq': 'Raw Download AA Seq'}),
|
| 667 |
+
raw_download,
|
| 668 |
+
on=['FusionGene','Raw Download AA Seq'],
|
| 669 |
+
how='inner'
|
| 670 |
+
)
|
| 671 |
+
test_merge_2 = test_merge_2.drop(columns=['Fold AA seq'])
|
| 672 |
+
test_merge_2['Seq Source'] = ['Webpage,Raw Download']*len(test_merge_2)
|
| 673 |
+
log_update(f"Merge on Webpage AA Seq and Raw Download AA Seq. len={len(test_merge_2)}")
|
| 674 |
+
|
| 675 |
+
test_merge = pd.concat([test_merge_1,test_merge_2])
|
| 676 |
+
test_merge['Len(AA seq)'] = test_merge['Raw Download AA Seq'].apply(lambda x: len(x))
|
| 677 |
+
# drop duplicates
|
| 678 |
+
test_merge = test_merge.drop_duplicates().reset_index(drop=True)
|
| 679 |
+
|
| 680 |
+
# for anything that has a CIF, keep the CIF
|
| 681 |
+
log_update(f"len test_merge before keeping CIFs over identical PDBs: {len(test_merge)}")
|
| 682 |
+
test_merge = test_merge.sort_values(by='Structure Type',ascending=True).reset_index(drop=True).groupby(['Hgene', 'Hchr', 'Hbp', 'Hstrand', 'Tgene', 'Tchr',
|
| 683 |
+
'Tbp', 'Tstrand', 'Len(AA seq)', 'FusionGene',
|
| 684 |
+
'Level', 'Raw Download AA Seq', 'pLDDT', 'pLDDTs','FusionGID', 'Seq Source']).agg(
|
| 685 |
+
{
|
| 686 |
+
'Structure Link': 'first',
|
| 687 |
+
'Structure Type': 'first'
|
| 688 |
+
}
|
| 689 |
+
).reset_index()
|
| 690 |
+
log_update(f"len after: {len(test_merge)}")
|
| 691 |
+
|
| 692 |
+
# for anything with multiple seq sources, concatenate them
|
| 693 |
+
log_update(f"len test_merge before combining seq sources: {len(test_merge)}")
|
| 694 |
+
test_merge = test_merge.groupby(['Structure Link','Hgene', 'Hchr', 'Hbp', 'Hstrand', 'Tgene', 'Tchr',
|
| 695 |
+
'Tbp', 'Tstrand', 'Len(AA seq)', 'FusionGene','Structure Type',
|
| 696 |
+
'Level', 'Raw Download AA Seq', 'pLDDT', 'pLDDTs', 'FusionGID', ]).agg(
|
| 697 |
+
{
|
| 698 |
+
'Seq Source': lambda x: ','.join(x)
|
| 699 |
+
}
|
| 700 |
+
).reset_index()
|
| 701 |
+
test_merge['Seq Source'] = test_merge['Seq Source'].apply(lambda x: ','.join(set(x.split(','))))
|
| 702 |
+
log_update(f"len after: {len(test_merge)}")
|
| 703 |
+
|
| 704 |
+
# are there cases of multiple folds for the same sequence? miraculously, yes!
|
| 705 |
+
dup_seqs = test_merge[test_merge['Raw Download AA Seq'].duplicated()]['Raw Download AA Seq'].unique().tolist()
|
| 706 |
+
|
| 707 |
+
# for anything with multiple folds / seq, randomly choose the first one.
|
| 708 |
+
log_update(f"len test_merge before randomly choosing first fold when one seq has multiple folds: {len(test_merge)}")
|
| 709 |
+
test_merge = test_merge.groupby(['Hgene', 'Hchr', 'Hbp', 'Hstrand', 'Tgene', 'Tchr',
|
| 710 |
+
'Tbp', 'Tstrand', 'Len(AA seq)', 'FusionGene',
|
| 711 |
+
'Level', 'Raw Download AA Seq', 'FusionGID', ]).agg(
|
| 712 |
+
{
|
| 713 |
+
'Structure Link': 'first',
|
| 714 |
+
'Structure Type': 'first',
|
| 715 |
+
'Seq Source': 'first',
|
| 716 |
+
'pLDDT': 'first',
|
| 717 |
+
'pLDDTs': 'first'
|
| 718 |
+
}
|
| 719 |
+
).reset_index()
|
| 720 |
+
log_update(f"len after: {len(test_merge)}")
|
| 721 |
+
|
| 722 |
+
# how many columns DO NOT have the right AlphaFold sequences?
|
| 723 |
+
source_str = test_merge['Seq Source'].value_counts().reset_index().rename(columns={'index': 'Seq Source','Seq Source': 'count'}).to_string(index=False)
|
| 724 |
+
source_str = "\t\t" + source_str.replace("\n","\n\t\t")
|
| 725 |
+
log_update(f"Distribution of sequence sources:\n{source_str}")
|
| 726 |
+
|
| 727 |
+
# dropping anything where AF sequence is wrong. Don't want to use these.
|
| 728 |
+
test_merge = test_merge.loc[test_merge['Seq Source'].str.contains('AlphaFold')].reset_index(drop=True)
|
| 729 |
+
log_update(f"Dropped rows where AlphaFold sequence was incorrect. New DataFrame length: {len(test_merge)}")
|
| 730 |
+
# make sure there's only one FusionGID number for each sequence for each GID
|
| 731 |
+
assert len(test_merge[test_merge.duplicated(['FusionGID','Raw Download AA Seq'])])==0
|
| 732 |
+
|
| 733 |
+
# round pLDDTs
|
| 734 |
+
test_merge['pLDDT'] = test_merge['pLDDT'].round(2)
|
| 735 |
+
|
| 736 |
+
# Finally, select only the columns we want
|
| 737 |
+
test_merge_v2 = test_merge[
|
| 738 |
+
['FusionGID', 'FusionGene', 'Raw Download AA Seq','Len(AA seq)', 'Hgene', 'Hchr', 'Hbp', 'Hstrand', 'Tgene', 'Tchr', 'Tbp', 'Tstrand',
|
| 739 |
+
'Level','Structure Link', 'Structure Type', 'pLDDT', 'pLDDTs', 'Seq Source']
|
| 740 |
+
].rename(
|
| 741 |
+
columns={
|
| 742 |
+
'Raw Download AA Seq': 'Fusion_Seq',
|
| 743 |
+
'Seq Source': 'Fusion_Seq_Source',
|
| 744 |
+
'Structure Link': 'Fusion_Structure_Link',
|
| 745 |
+
'Structure Type': 'Fusion_Structure_Type',
|
| 746 |
+
'pLDDT': 'Fusion_pLDDT',
|
| 747 |
+
'pLDDTs': 'Fusion_AA_pLDDTs',
|
| 748 |
+
'Len(AA seq)': 'Fusion_Length'
|
| 749 |
+
}
|
| 750 |
+
)
|
| 751 |
+
log_update(f"Unique FusionGIDs: {len(test_merge_v2['FusionGID'].unique())}")
|
| 752 |
+
log_update(f"Number of structures: {len(test_merge_v2)}")
|
| 753 |
+
|
| 754 |
+
# Note that test_merge_v2 will still have duplicates where it's same seq, different ID
|
| 755 |
+
log_update("\nChecking for duplicate sequences..")
|
| 756 |
+
log_update(f"\tThe structure-based fusion database of length {len(test_merge_v2)} has {len(test_merge_v2['Fusion_Seq'].unique())} unique fusion sequences.")
|
| 757 |
+
dup_seqs = test_merge_v2[test_merge_v2['Fusion_Seq'].duplicated()]['Fusion_Seq'].tolist()
|
| 758 |
+
dup_seqs_df = test_merge_v2.loc[test_merge_v2['Fusion_Seq'].isin(dup_seqs)].reset_index(drop=True)
|
| 759 |
+
dup_seqs_df['FusionGID'] = dup_seqs_df['FusionGID'].astype(str)
|
| 760 |
+
dup_seqs_df = dup_seqs_df.groupby('Fusion_Seq').agg({
|
| 761 |
+
'FusionGID': lambda x: ','.join(x),
|
| 762 |
+
'FusionGene': lambda x: ','.join(x)
|
| 763 |
+
})
|
| 764 |
+
dup_seqs_df_str = dup_seqs_df.to_string(index=False)
|
| 765 |
+
dup_seqs_df_str = "\t"+dup_seqs_df_str.replace("\n","\n\t")
|
| 766 |
+
log_update(f"\tShowing FUsionGIDs and FusionGenes for duplicated sequences below:\n{dup_seqs_df_str}")
|
| 767 |
+
|
| 768 |
+
# round pLDDT column
|
| 769 |
+
heads_tails_analyzed['Avg pLDDT'] = heads_tails_analyzed['Avg pLDDT'].round(2)
|
| 770 |
+
# merge treating data as head data
|
| 771 |
+
level_2_3_v2 = pd.merge(
|
| 772 |
+
level_2_3,
|
| 773 |
+
heads_tails_analyzed.rename(columns={'UniProtID': 'HGUniProtAcc', 'Avg pLDDT': 'HG_pLDDT', 'All pLDDTs': 'HG_AA_pLDDTs', 'Seq': 'HG_Seq'}),
|
| 774 |
+
on='HGUniProtAcc',
|
| 775 |
+
how='left'
|
| 776 |
+
)
|
| 777 |
+
# merge treating data as tail data
|
| 778 |
+
level_2_3_v2 = pd.merge(
|
| 779 |
+
level_2_3_v2,
|
| 780 |
+
heads_tails_analyzed.rename(columns={'UniProtID': 'TGUniProtAcc', 'Avg pLDDT': 'TG_pLDDT', 'All pLDDTs': 'TG_AA_pLDDTs', 'Seq': 'TG_Seq'}),
|
| 781 |
+
on='TGUniProtAcc',
|
| 782 |
+
how='left'
|
| 783 |
+
)
|
| 784 |
+
|
| 785 |
+
# giant_level2_3 with valid structures only, no duplicate sequences, and head and tail proteins' uniprot IDs, pLDDTs, and sequences
|
| 786 |
+
test_merge_v2.to_csv(f'processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv',index=False)
|
| 787 |
+
log_update("Saved file with all fusion structure pLDDTs to: processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv")
|
| 788 |
+
|
| 789 |
+
# level_2_3 with the head and tail proteins' uniprot IDs, pLDDTs, and sequences
|
| 790 |
+
level_2_3_v2.to_csv(f'processed_data/fusionpdb/FusionPDB_level2-3_cleaned_FusionGID_info.csv',index=False)
|
| 791 |
+
log_update("Saved file with all fusion protein heads and tails, and their structure pLDDTs to: processed_data/fusionpdb/FusionPDB_level2-3_cleaned_FusionGID_info.csv")
|
| 792 |
+
|
| 793 |
+
def main():
|
| 794 |
+
with open_logfile("process_fusion_structures_log.txt"):
|
| 795 |
+
process_fusions_and_hts()
|
| 796 |
+
|
| 797 |
+
if __name__ == "__main__":
|
| 798 |
+
main()
|
| 799 |
+
|
fuson_plm/benchmarking/caid/processed_data/CAID-2_Disorder_NOX_Processed.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:8a6947f68c9d85ddaf7888207dde0d75a55a6e542da7a67aa5f3eefcf62a1511
|
| 3 |
+
size 324778
|
fuson_plm/benchmarking/caid/processed_data/IDP-CRF_Training_Dataset.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:1f575340f3c68db5b5dcf1734301c946fa2d3aacb6583bcfca53e0610604553f
|
| 3 |
+
size 2966294
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/AlphaFold-disorder_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:0ec1ffc27bddbb7a456c66391845f9487477077ebbcb99537310a8ae224ead78
|
| 3 |
+
size 977219
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/AlphaFold-rsa_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:8f63fa90e6a629a9a64f61e9409e33af2aa134e026197f91fbef3c75b1574d38
|
| 3 |
+
size 977736
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/DISOPRED3-diso_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:5334217cd5290148af34134a1417c13fc88464ea757de5ccdc89fde9110b26b4
|
| 3 |
+
size 1772014
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/DeepIDP-2L_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:268038cb09ff3363dfa6322f547ff00cfb40b297e465e92fc281357d62df3215
|
| 3 |
+
size 1772014
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/DisoPred_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:952bc241621271e642da19abacc245899801733d7fcd16db066e19ab1f716ef0
|
| 3 |
+
size 1687118
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/Dispredict3_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:a6a7293ebd84a6430c399e950a4d56adbb208a9f0ca568c32a117d78c47553e7
|
| 3 |
+
size 1772014
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/ESpritz-D_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:9da4c18279b48542e6da32e4306665f3712d0eeb37d8028323adc1058f113ac2
|
| 3 |
+
size 1772014
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/IDP-Fusion_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:a15182f4cb34d78c68555904b9d1fb5a25a10a425ce9414647c2d2057ebe92a6
|
| 3 |
+
size 1742594
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/IUPred3_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:e84fbaeb072f3e10cd2c939f5022bdaa196061651463aadd7ad526f65d5b707a
|
| 3 |
+
size 1772018
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/disomine_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:9a13376dcbbb0858ab091d2694a40a34bcbf99937a4faa9b70fcd61ee3a6984d
|
| 3 |
+
size 1449978
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPlr2_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:66420600ab69289e2a1a1452b12936c9b289b7e922571eb6efb139b16eab4db7
|
| 3 |
+
size 1736172
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPnn2_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:c8de27d681d8c216a2a430ace8bfa130f106d4174fb066deec8f9e780e04f51c
|
| 3 |
+
size 1736172
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPnn_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:abb9d0b9cceae039b17105f9baaf023dd6e0d0da7aa48bece988cdcc5f7928c3
|
| 3 |
+
size 1772014
|
fuson_plm/benchmarking/caid/processed_data/caid2_competition_results/flDPtr_CAID-2_Disorder_NOX.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:27385d5a1d9348fdd90f1cfb516cd36209f61d0fe113439be8f39113825b3a03
|
| 3 |
+
size 1736172
|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EML4-ALK.png
ADDED

|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EML4::ALK_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:f3d9a65f1e8ba25cdad55f016bcde23f739c9526f29983c30579e7a49fcd409e
|
| 3 |
+
size 404717
|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EWSR1-FLI1.png
ADDED

|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_EWSR1::FLI1_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:8043d8bc2652721158e7f0cd8d40a94951ed85b781b9af9a997d0c961c022d29
|
| 3 |
+
size 77468
|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_PAX3-FOXO1.png
ADDED

|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_PAX3::FOXO1_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:93546673955907592808d58145040b075bf6dd58d08eebc412ef53d23e060858
|
| 3 |
+
size 30322
|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_SS18-SSX1.png
ADDED

|
fuson_plm/benchmarking/caid/processed_data/figures/fusion_disorder/plddt_sequence_SS18::SSX1_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:86380dc4bb6c213591550b659d4e4dbb11650b8d31c0a1c32f1ca0d02e90abd9
|
| 3 |
+
size 61266
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/disorder_nox_histogram.png
ADDED

|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/disorder_nox_histogram_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:32c2dfea9a8f45cc1d4005c7aac2fd729f2d930a8d68b17e05dc92175e85bead
|
| 3 |
+
size 3051
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/fusions_histogram.png
ADDED
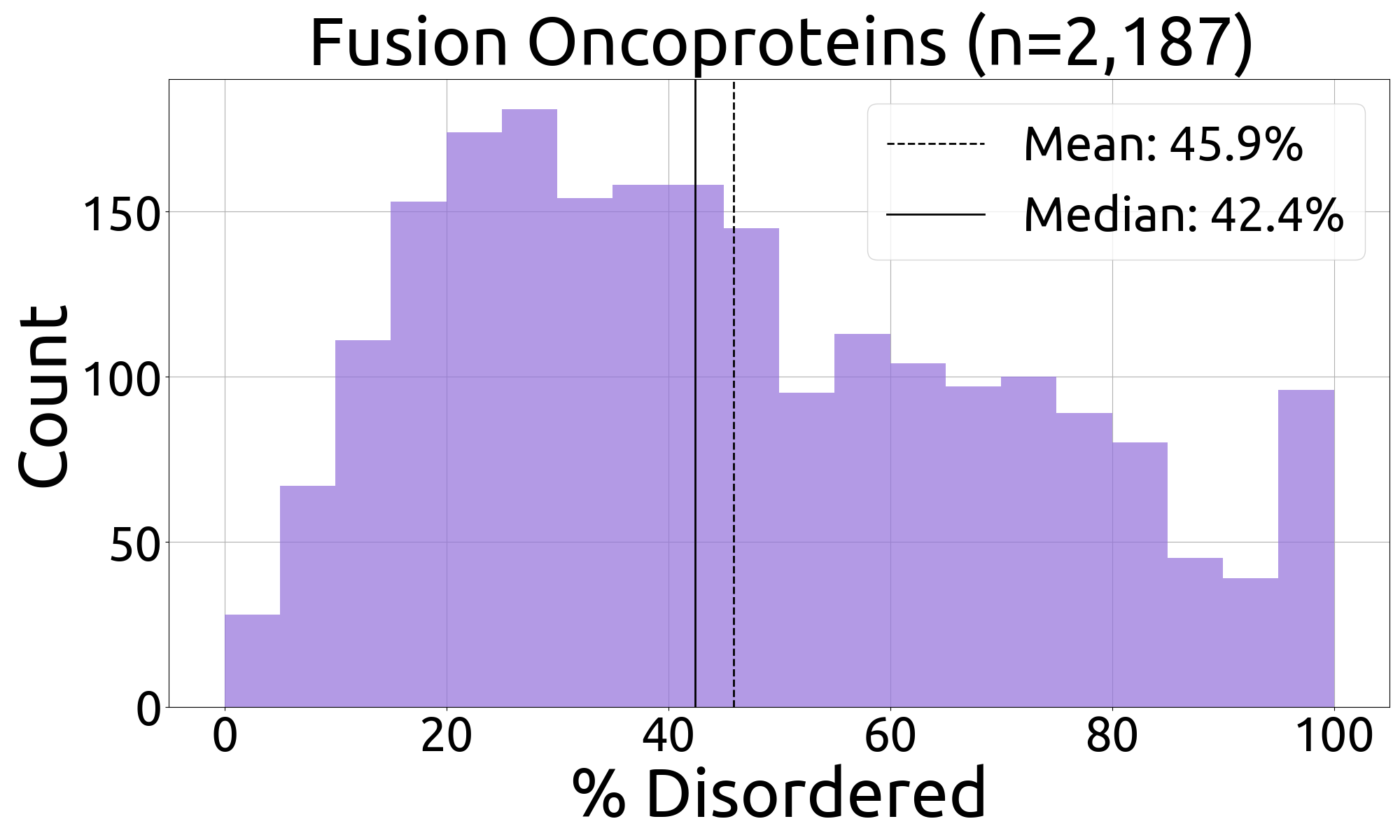
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/fusions_histogram_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:a3f0a468334748fe08f1a230ea0897d783bdaf52b4674a3469c4d4111cbcaf98
|
| 3 |
+
size 34084
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/heads_histogram.png
ADDED
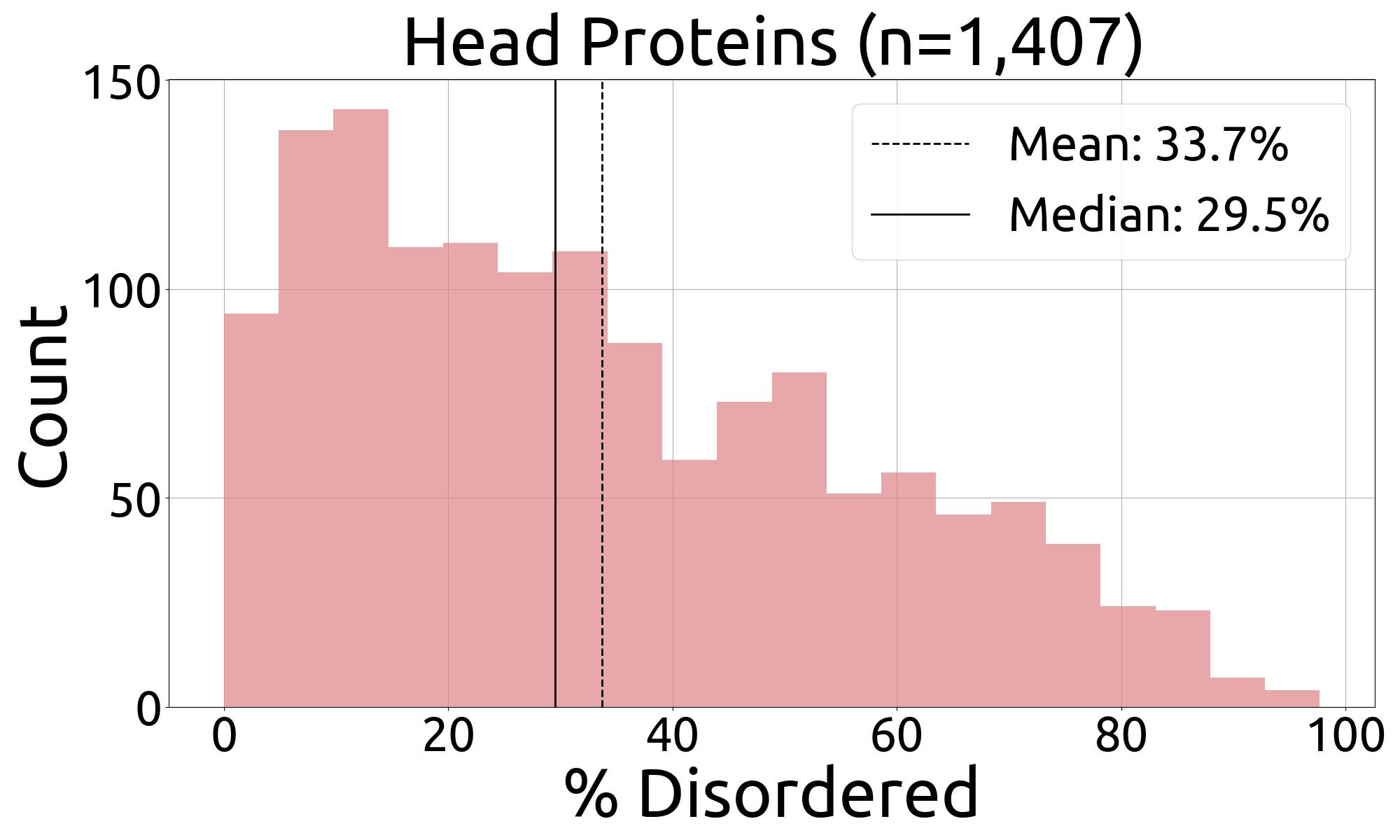
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/heads_histogram_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:3c3c119114f8f3a98e8f16bb9b531102e4fa61b7cfe2eea1e641439d7b4c9762
|
| 3 |
+
size 19279
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/tails_histogram.png
ADDED
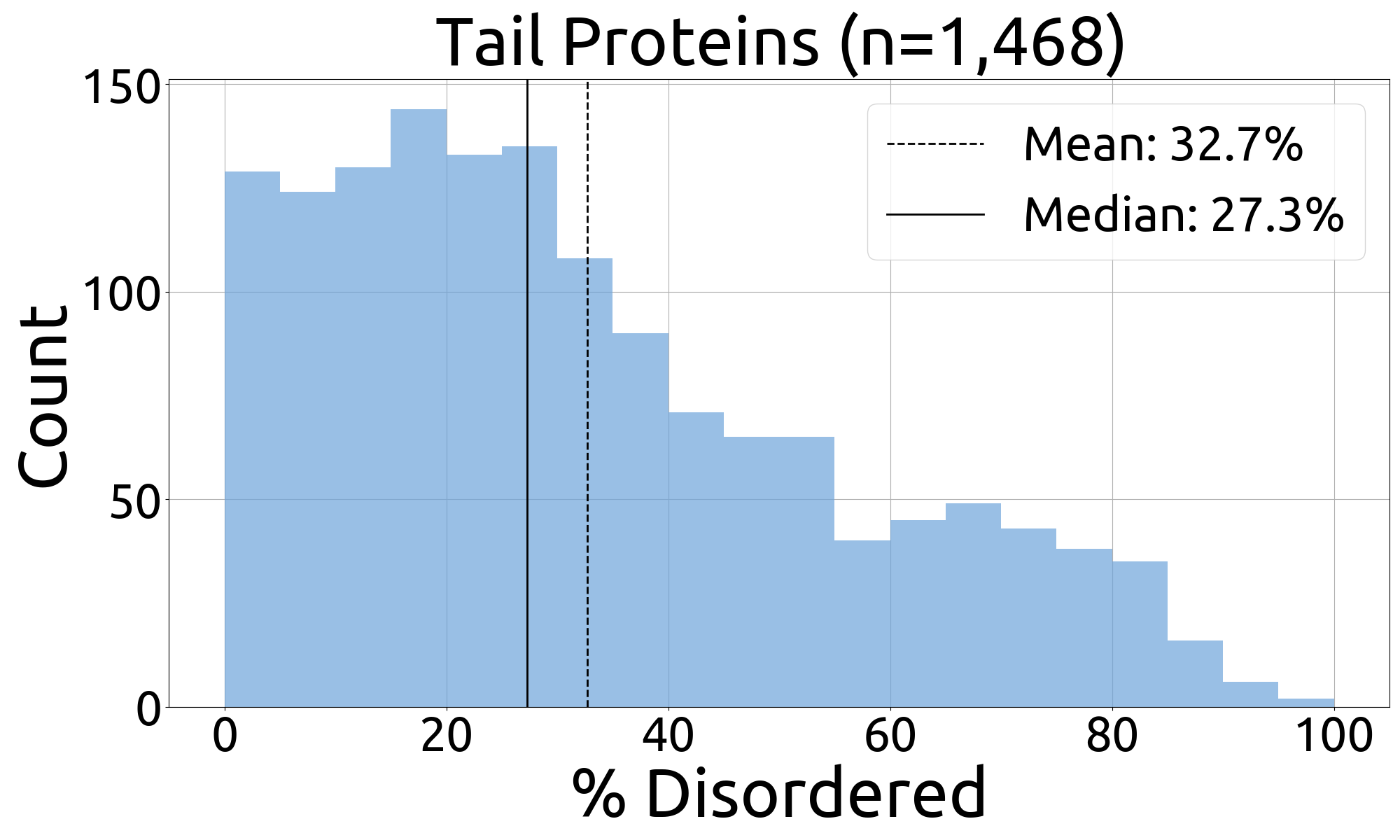
|
fuson_plm/benchmarking/caid/processed_data/figures/histograms/tails_histogram_source_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:cbce7586f8f1a39696f6562c482aa6541cc3c99b104a207278922a47ba0a8dab
|
| 3 |
+
size 20101
|
fuson_plm/benchmarking/caid/processed_data/flDPnn_Training_Dataset.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:9882e290f110d47282ae81451a2e073ff2f579a957baa828144ca604f91f191e
|
| 3 |
+
size 447048
|
fuson_plm/benchmarking/caid/processed_data/flDPnn_Validation_Dataset.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:6568731760f2cb9daf20f55fec0014e0ee3eeaaa32c929770a16275792c837d6
|
| 3 |
+
size 61366
|
fuson_plm/benchmarking/caid/processed_data/fusionpdb/FusionPDB_level2-3_cleaned_FusionGID_info.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:aae5d5e39c4c52a1d07f0c427177004692688d0d8f447fd6b4eb83b0adbad6f3
|
| 3 |
+
size 24073232
|
fuson_plm/benchmarking/caid/processed_data/fusionpdb/FusionPDB_level2-3_cleaned_structure_info.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:0e93f109073115fddbc97d493674ad2acc515b3c37bc62db23461d2a3b469e0f
|
| 3 |
+
size 13726330
|
fuson_plm/benchmarking/caid/processed_data/fusionpdb/fusion_heads_and_tails.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:61e8a29ba88d431c54ac40def2a1551ce21c203c2f9bb2f49a87b14ba0b41493
|
| 3 |
+
size 68031
|
fuson_plm/benchmarking/caid/processed_data/fusionpdb/heads_tails_structural_data.csv
ADDED
|
@@ -0,0 +1,3 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
version https://git-lfs.github.com/spec/v1
|
| 2 |
+
oid sha256:9b6115ec413dccb68dfe3c60c239c625a42161be6b87be14f2b3c1566452587e
|
| 3 |
+
size 15471991
|
fuson_plm/benchmarking/caid/raw_data/caid2_competition_results/AlphaFold-disorder.caid
ADDED
|
The diff for this file is too large to render.
See raw diff
|
|
|
fuson_plm/benchmarking/caid/raw_data/caid2_competition_results/AlphaFold-rsa.caid
ADDED
|
The diff for this file is too large to render.
See raw diff
|
|
|